
Complete Chloroplast Genomes of Four Oaks from the Section Cyclobalanopsis Improve the Phylogenetic Analysis and Understanding of Evolutionary Processes in the Genus Quercus
 ,
,  , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Samples and DNA Extraction and Sequencing
2.2. Chloroplast Genome Assembly, Annotation, and Visualization
2.3. Repeated Sequence Analysis
2.4. Codon Usage Bias Analysis
2.5. Comparative Genome Analyses of Chloroplast Genomes
2.6. Phylogenetic Analysis
2.7. Evolutionary Selection Pressure Analysis
3. Results
3.1. Chloroplast Genome Structures and Features of the Four Quercus Section Cyclobalanopsis Species
3.2. Repeated Sequences Analysis of Four Quercus Section Cyclobalanopsis Species
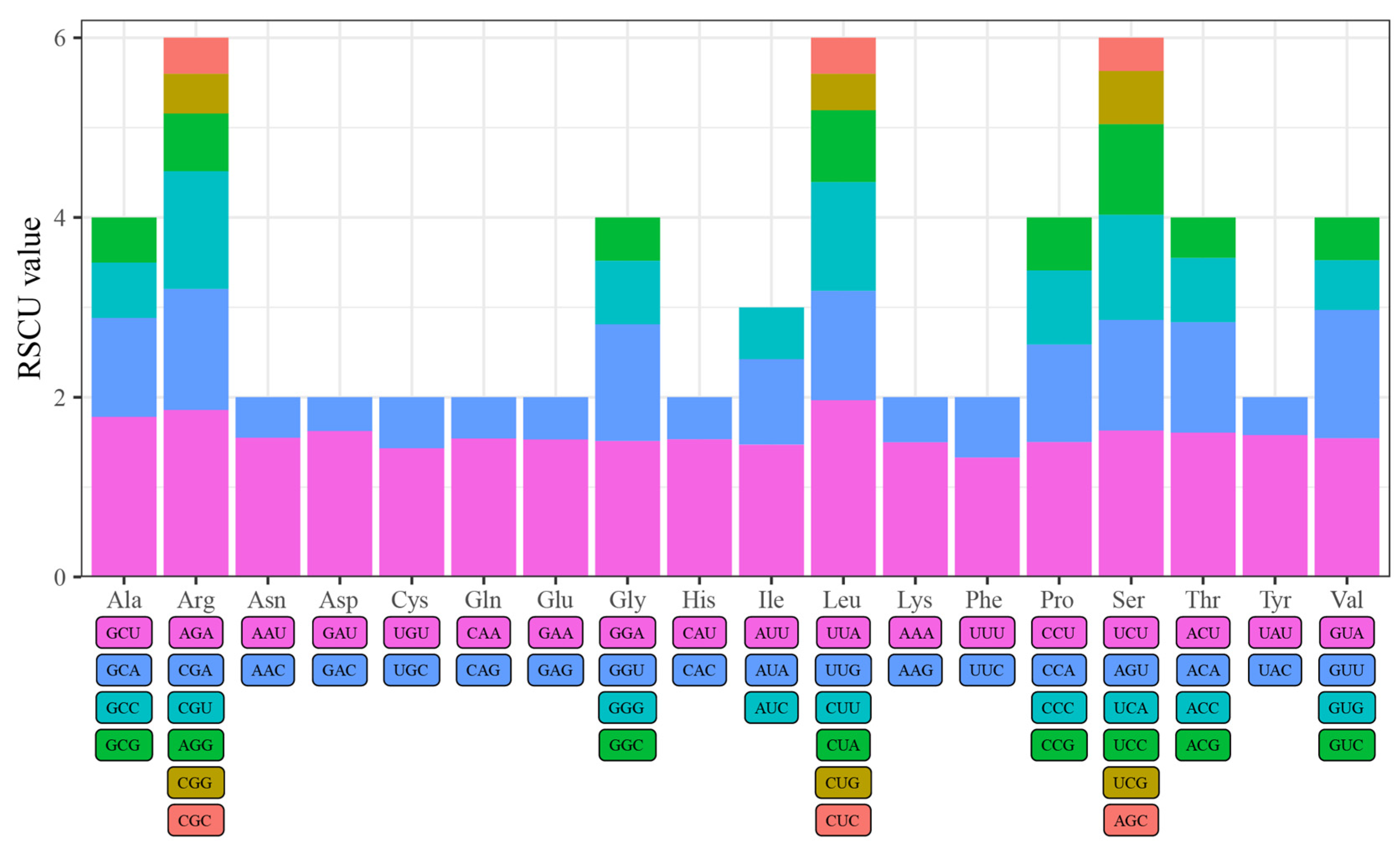
3.3. Codon Usage Bias Analysis of Four Quercus Section Cyclobalanopsis Species
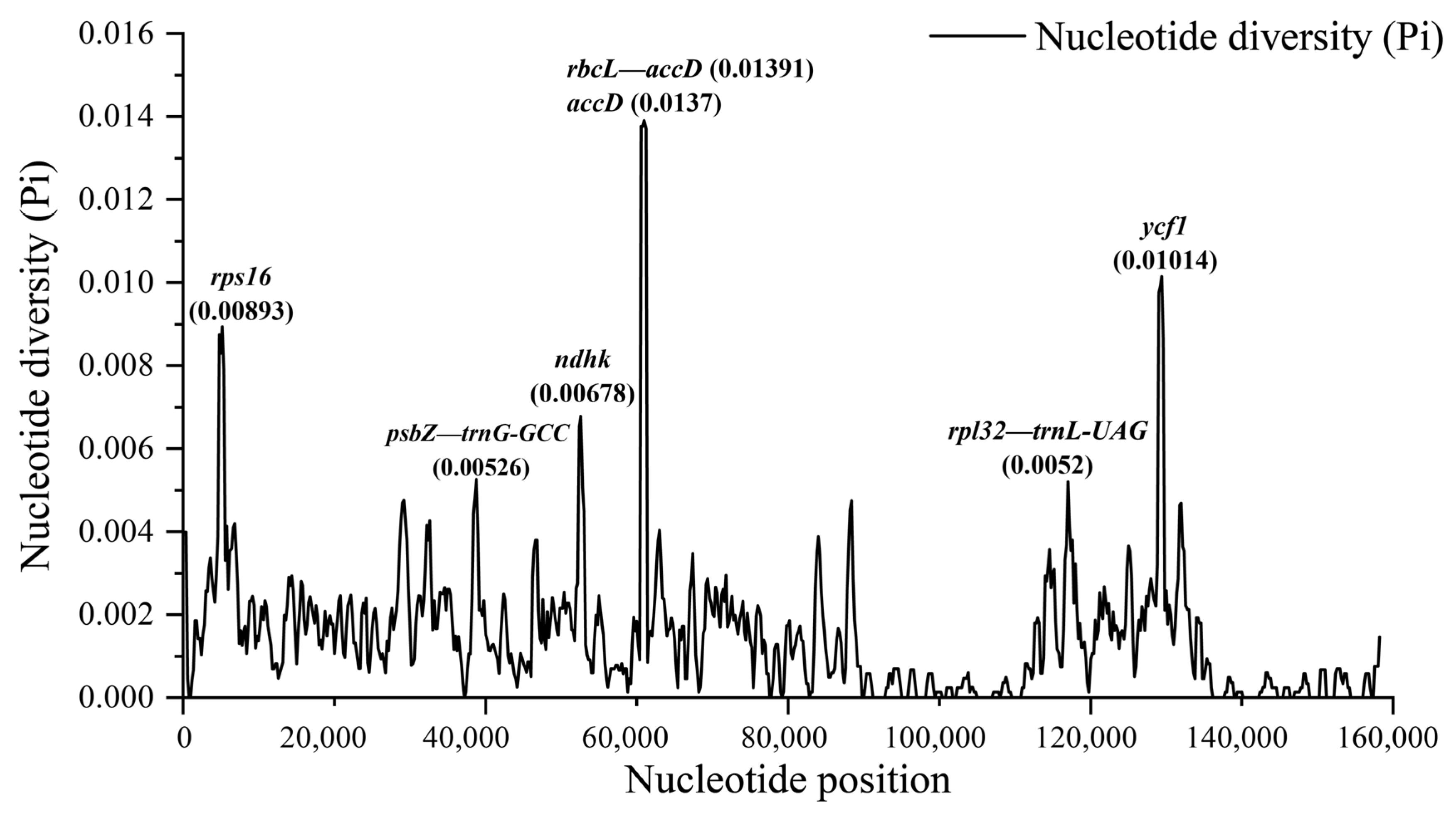
3.4. Comparative Genome of Chloroplast Genomes of Quercus Section Cyclobalanopsis
3.5. Phylogenetic Relationships
3.6. Selection Pressure Analysis
4. Discussion
4.1. Architecture of Chloroplast Genomes in Quercus Section Cyclobalanopsis
4.2. Phylogeny and Evolution of the Quercus Chloroplast Genome
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Harlan, J.R. Our Vanishing Genetic Resources: Modern varieties replace ancient populations that have provided genetic variability for plant breeding programs. Science 1975, 188, 618–621. [Google Scholar] [CrossRef]
- Hoban, S.; Bruford, M.; Jackson, J.D.; Lopes-Fernandes, M.; Heuertz, M.; Hohenlohe, P.A.; Paz-Vinas, I.; Sjögren-Gulve, P.; Segelbacher, G.; Vernesi, C.; et al. Genetic diversity targets and indicators in the CBD post-2020 Global Biodiversity Framework must be improved. Conserv. Genet. 2020, 248, 108654. [Google Scholar] [CrossRef]
- Stange, M.; Barrett, R.D.; Hendry, A.P. The importance of genomic variation for biodiversity, ecosystems and people. Nat. Rev. Genet. 2021, 22, 89–105. [Google Scholar] [CrossRef] [PubMed]
- Hoban, S.; Archer, F.I.; Bertola, L.D.; Bragg, J.G.; Breed, M.F.; Bruford, M.W.; Coleman, M.A.; Ekblom, R.; Funk, W.C.; Grueber, C.E.; et al. Global genetic diversity status and trends: Towards a suite of Essential Biodiversity Variables (EBVs) for genetic composition. Biol. Rev. Camb. Philos. Soc. 2022, 97, 1511–1538. [Google Scholar] [CrossRef] [PubMed]
- Douglas, S.E. Chloroplast origins and evolution. In The Molecular Biology of Cyanobacteria; Springer: Dordrecht, The Netherlands, 1994; pp. 91–118. [Google Scholar]
- Jiang, H.; Tian, J.; Yang, J.X.; Dong, X.; Zhong, Z.X.; Mwachala, G.; Zhang, C.F.; Hu, G.W.; Wang, Q.F. Comparative and phylogenetic analyses of six Kenya Polystachya (Orchidaceae) species based on the complete chloroplast genome sequences. BMC Plant Biol. 2022, 22, 177. [Google Scholar] [CrossRef] [PubMed]
- Li, H.-T.; Yi, T.-S.; Gao, L.-M.; Ma, P.-F.; Zhang, T.; Yang, J.-B.; Gitzendanner, M.A.; Fritsch, P.W.; Cai, J.; Luo, Y.; et al. Origin of angiosperms and the puzzle of the Jurassic gap. Nat. Plants 2019, 5, 461–470. [Google Scholar] [CrossRef] [PubMed]
- Li, H.-T.; Luo, Y.; Gan, L.; Ma, P.-F.; Gao, L.-M.; Yang, J.-B.; Cai, J.; Gitzendanner, M.A.; Fritsch, P.W.; Zhang, T.; et al. Plastid phylogenomic insights into relationships of all flowering plant families. BMC Biol. 2021, 19, 232. [Google Scholar] [CrossRef] [PubMed]
- Birky, C.W., Jr. Uniparental inheritance of mitochondrial and chloroplast genes: Mechanisms and evolution. Proc. Natl. Acad. Sci. USA 1995, 92, 11331–11338. [Google Scholar] [CrossRef] [PubMed]
- Hodel, R.G.; Knowles, L.L.; McDaniel, S.F.; Payton, A.C.; Dunaway, J.F.; Soltis, P.S.; Soltis, D.E. Terrestrial species adapted to sea dispersal: Differences in propagule dispersal of two Caribbean mangroves. Mol. Ecol. 2018, 27, 4612–4626. [Google Scholar] [CrossRef]
- Nock, C.J.; Baten, A.; King, G.J. Complete chloroplast genome of Macadamia integrifoliaconfirms the position of the Gondwanan early-diverging eudicot family Proteaceae. BMC Genom. 2014, 15, S13. [Google Scholar] [CrossRef]
- Ramsey, A.J.; Mandel, J.R. When one genome is not enough: Organellar heteroplasmy in plants. Annu. Plant Rev. Online 2018, 2, 619–658. [Google Scholar] [CrossRef]
- Denk, T.; Grimm, G.W.; Manos, P.S.; Deng, M.; Hipp, A.L. An Updated Infrageneric Classification of the Oaks: Review of Previous Taxonomic Schemes and Synthesis of Evolutionary Patterns. In Oaks Physiological Ecology. Exploring the Functional Diversity of Genus Quercus L.; Gil-Pelegrin, E., Peguero-Pina, J., Eds.; Tree Physiology Book Series; Springer: Cham, Switzerland, 2017; Volume 7, pp. 13–38. [Google Scholar]
- Jin, D.M.; Yuan, Q.; Dai, X.L.; Kozlowski, G.; Song, Y.G. Enhanced precipitation has driven the evolution of subtropical evergreen broad-leaved forests in eastern China since the early Miocene: Evidence from ring-cupped oaks. J. Syst. Evol. 2023. ahead of print. [Google Scholar] [CrossRef]
- Carrero, C.; Jerome, D.; Beckman, E.; Byrne, A.; Coombes, A.J.; Deng, M.; González-Rodríguez, A.; Hoang, V.S.; Khoo, E.; Nguyen, N.; et al. The Red List of Oaks 2020; The Morton Arboretum: Lisle, IL, USA, 2020; p. 5. [Google Scholar]
- Manos, P.S.; Zhou, Z.K.; Cannon, C.H. Systematics of Fagaceae: Phylogenetic tests of reproductive trait evolution. Int. J. Plant Sci. 2001, 162, 1361–1379. [Google Scholar] [CrossRef]
- Denk, T.; Grimm, G.W. The oaks of western Eurasia: Traditional classifications and evidence from two nuclear markers. Taxon 2010, 59, 351–366. [Google Scholar] [CrossRef]
- Deng, M.; Zhou, Z.K.; Li, Q.S. Taxonomy and systematics of Quercus subgenus Cyclobalanopsis. Int. Oaks 2013, 24, 48–60. [Google Scholar]
- Xu, J.; Deng, M.; Jiang, X.L.; Westwood, M.; Song, Y.G.; Turkington, R. Phylogeography of Quercus glauca (Fagaceae), a dominant tree of East Asian subtropical evergreen forests, based on three chloroplast DNA interspace sequences. Tree Genet. Genomes 2015, 11, 805. [Google Scholar] [CrossRef]
- Xu, J.; Jiang, X.L.; Deng, M.; Westwood, M.; Song, Y.G.; Zheng, S.S. Conservation genetics of rare trees restricted to subtropical montane cloud forests in southern China: A case study from Quercus arbutifolia (Fagaceae). Tree Genet. Genomes 2016, 12, 90. [Google Scholar] [CrossRef]
- An, M.; Deng, M.; Zheng, S.S.; Jiang, X.L.; Song, Y.G. Introgression threatens the genetic diversity of Quercus austrocochinchinensis (Fagaceae), an endangered oak: A case inferred by molecular markers. Front. Plant Sci. 2017, 8, 229. [Google Scholar] [CrossRef] [PubMed]
- Deng, M.; Jiang, X.L.; Hipp, A.L.; Manos, P.S.; Hahn, M. Phylogeny and biogeography of East Asian evergreen oaks (Quercus section Cyclobalanopsis; Fagaceae): Insights into the Cenozoic history of evergreen broad-leaved forests in subtropical Asia. Mol. Phylogenetics Evol. 2018, 119, 170–181. [Google Scholar] [CrossRef]
- Xu, J.; Song, Y.G.; Deng, M.; Jiang, X.L.; Zheng, S.S.; Li, Y. Seed Germination Schedule and Environmental Context Shaped the Population Genetic Structure of Subtropical Evergreen Oaks on the Yun-Gui Plateau, Southwest China. Heredity 2020, 124, 499–513. [Google Scholar] [CrossRef]
- Yang, Y.; Zhou, T.; Qian, Z.; Zhao, G. Phylogenetic relationships in Chinese oaks (Fagaceae, Quercus): Evidence from plastid genome using low-coverage whole genome sequencing. Genomics 2021, 113, 1438–1447. [Google Scholar] [CrossRef]
- Li, Y.; Wang, T.R.; Kozlowski, G.; Liu, M.H.; Yi, L.T.; Song, Y.G. Complete chloroplast genome of an endangered species Quercus litseoides, and its comparative, evolutionary, and phylogenetic study with other Quercus section Cyclobalanopsis species. Genes 2022, 13, 1184. [Google Scholar] [CrossRef]
- Li, X.; Li, Y.; Zang, M.; Li, M.; Fang, Y. Complete chloroplast genome sequence and phylogenetic analysis of Quercus acutissima. Int. J. Mol. Sci. 2018, 19, 2443. [Google Scholar] [CrossRef]
- Wang, T.R.; Wang, Z.W.; Song, Y.G.; Kozlowski, G. The complete chloroplast genome sequence of Quercus ningangensis and its phylogenetic implication. Plant Fungal Syst. 2021, 66, 155–165. [Google Scholar] [CrossRef]
- Wei, R.; Li, Q. The complete chloroplast genome of endangered species Stemona parviflora: Insight into the phylogenetic relationship and conservation implications. Genes 2022, 13, 1361. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.-S.; Yang, J.; Hu, H.-L.; Xia, R.-X.; Li, Y.-P.; Su, J.-F.; Li, Q.; Liu, Y.-Q.; Qin, L. A high level of chloroplast genome sequence variability in the Sawtooth Oak Quercus acutissima. Int. J. Biol. Macromol. 2020, 152, 340–348. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Zhou, T.; Duan, D.; Yang, J.; Feng, L.; Zhao, G. Comparative Analysis of the Complete Chloroplast Genomes of Five Quercus Species. Front. Plant Sci. 2016, 7, 959. [Google Scholar] [CrossRef] [PubMed]
- Doyle, J.J.; Doyle, J.L. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem. Bull. 1987, 19, 11–15. [Google Scholar]
- Batzoglou, S.; Berger, B.; Mesirov, J.; Lander, E.S. Sequencing a genome by walking with clone-end sequences: A mathematical analysis. Genome Res. 1999, 9, 1163–1174. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.J.; Yu, W.B.; Yang, J.B.; Song, Y.; DePamphilis, C.W.; Yi, T.S.; Li, D.Z. GetOrganelle: A fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome Biol. 2020, 21, 241. [Google Scholar] [CrossRef] [PubMed]
- Wick, R.R.; Schultz, M.B.; Zobel, J.; Holt, K.E. Bandage: Interactive visualization of de novo genome assemblies. Bioinformatics 2015, 31, 3350–3352. [Google Scholar] [CrossRef] [PubMed]
- Tillich, M.; Lehwark, P.; Pellizzer, T.; Ulbricht-Jones, E.S.; Fischer, A.; Bock, R.; Greiner, S. GeSeq–versatile and accurate annotation of organelle genomes. Nucleic Acids Res. 2017, 45, W6–W11. [Google Scholar] [CrossRef] [PubMed]
- Lohse, M.; Drechsel, O.; Kahlau, S.; Bock, R. OrganellarGenomeDRAW—A suite of tools for generating physical maps of plastid and mitochondrial genomes and visualizing expression data sets. Nucleic Acids Res. 2013, 41, W575–W581. [Google Scholar] [CrossRef] [PubMed]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Drummond, A. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef] [PubMed]
- Beier, S.; Thiel, T.; Münch, T.; Scholz, U.; Mascher, M. MISA-web: A web server for microsatellite prediction. Bioinformatics 2017, 33, 2583–2585. [Google Scholar] [CrossRef]
- Kurtz, S.; Choudhuri, J.V.; Ohlebusch, E.; Schleiermacher, C.; Stoye, J.; Giegerich, R. REPuter: The manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 2001, 29, 4633–4642. [Google Scholar] [CrossRef]
- Benson, G. Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res. 1999, 27, 573–580. [Google Scholar] [CrossRef]
- Liang, C.; Wang, L.; Lei, J.; Duan, B.; Ma, W.; Xiao, S.; Qi, H.; Wang, Z.; Liu, Y.; Shen, X.; et al. A comparative analysis of the chloroplast genomes of four Salvia medicinal plants. Engineering 2019, 5, 907–915. [Google Scholar] [CrossRef]
- Gribskov, M.; Devereux, J.; Burgess, R.R. The codon preference plot: Graphic analysis of protein coding sequences and prediction of gene expression. Nucleic Acids Res. 1984, 12, 539–549. [Google Scholar] [CrossRef] [PubMed]
- Sharp, P.M.; Li, W.H. The codon adaptation index-a measure of directional synonymous codon usage bias, and its potential applications. Nucleic Acids Res. 1987, 15, 1281–1295. [Google Scholar] [CrossRef]
- Amiryousefi, A.; Hyvönen, J.; Poczai, P. IRscope: An online program to visualize the junction sites of chloroplast genomes. Bioinformatics 2018, 34, 3030–3031. [Google Scholar] [CrossRef] [PubMed]
- Brudno, M.; Malde, S.; Poliakov, A.; Do, C.B.; Couronne, O.; Dubchak, I.; Batzoglou, S. Glocal alignment: Finding rearrangements during alignment. Bioinformatics 2003, 19 (Suppl. 1), i54–i62. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Rozas, J.; Ferrer-Mata, A.; Sánchez-DelBarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sánchez-Gracia, A. DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef] [PubMed]
- Gou, W.; Jia, S.B.; Price, M.; Guo, X.L.; Zhou, S.D.; He, X.J. Complete plastid genome sequencing of eight species from Hansenia, Haplosphaera and Sinodielsia (Apiaceae): Comparative analyses and phylogenetic implications. Plants 2020, 9, 1523. [Google Scholar] [CrossRef] [PubMed]
- Guindon, S.; Dufayard, J.F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.T.; Schmidt, H.A.; Von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Yang, Z. Estimating the pattern of nucleotide substitution. J. Mol. Evol. 1994, 39, 105–111. [Google Scholar] [CrossRef]
- Hurst, L.D. The Ka/Ks ratio: Diagnosing the form of sequence evolution. Trends Genet. 2002, 18, 486–487. [Google Scholar] [CrossRef]
- Yang, Z. PAML 4: Phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 2007, 24, 1586–1591. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Nielsen, R. Codon-substitution models for detecting molecular adaptation at individual sites along specific lineages. Mol. Biol. Evol. 2002, 19, 908–917. [Google Scholar] [CrossRef]
- Anisimova, M.; Gascuel, O. Approximate likelihood-ratio test for branches: A fast, accurate, and powerful alternative. Syst. Biol. 2006, 55, 539–552. [Google Scholar] [CrossRef]
- Wicke, S.; Schneeweiss, G.M.; Depamphilis, C.W.; Müller, K.F.; Quandt, D. The evolution of the plastid chromosome in land plants: Gene content, gene order, gene function. Plant Mol. Biol. 2011, 76, 273–297. [Google Scholar] [CrossRef] [PubMed]
- Yin, K.; Zhang, Y.; Li, Y.; Du, F.K. Different natural selection pressures on the atpF gene in evergreen sclerophyllous and deciduous oak species: Evidence from comparative analysis of the complete chloroplast genome of Quercus aquifolioides with other oak species. Int. J. Mol. Sci. 2018, 19, 1042. [Google Scholar] [CrossRef] [PubMed]
- Zong, D.; Zhou, A.; Zhang, Y.; Zou, X.; Li, D.; Duan, A.; He, C. Characterization of the complete chloroplast genomes of five Populus species from the western Sichuan plateau, southwest China: Comparative and phylogenetic analyses. PeerJ 2019, 7, e6386. [Google Scholar] [CrossRef] [PubMed]
- Maréchal, A.; Brisson, N. Recombination and the maintenance of plant organelle genome stability. New Phytol. 2010, 186, 299–317. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.; Penaflor, C.; Kuehl, J.V.; Leebens-Mack, J.; E Carlson, J.; Depamphilis, C.W.; Boore, J.L.; Jansen, R.K. Complete Plastid Genome Sequences of Drimys, Liriodendron, and Piper: Implications for the Phylogenetic Relationships of Magnoliids. BMC Evol. Biol. 2006, 6, 77. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.; Lee, S.-C.; Lee, J.; Yu, Y.; Yang, K.; Choi, B.-S.; Koh, H.-J.; Waminal, N.E.; Choi, H.-I.; Kim, N.-H.; et al. Complete chloroplast and ribosomal sequences for 30 accessions elucidate evolution of Oryza AA genome species. Sci. Rep. 2015, 5, 15655. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Chang, E.M.; Liu, J.F.; Huang, Y.N.; Wang, Y.; Yao, N.; Jiang, Z.P. Complete chloroplast genome sequence and phylogenetic analysis of Quercus bawanglingensis Huang, Li et Xing, a vulnerable oak tree in China. Forests 2019, 10, 587. [Google Scholar] [CrossRef]
- Novák, P.; Guignard, M.S.; Neumann, P.; Kelly, L.J.; Mlinarec, J.; Koblížková, A.; Leitch, A.R. Repeat-sequence turnover shifts fundamentally in species with large genomes. Nat. Plants 2020, 6, 1325–1329. [Google Scholar] [CrossRef]
- Weng, M.L.; Blazier, J.C.; Govindu, M.; Jansen, R.K. Reconstruction of the ancestral plastid genome in Geraniaceae reveals a correlation between genome rearrangements, repeats, and nucleotide substitution rates. Mol. Biol. Evol. 2014, 31, 645–659. [Google Scholar] [CrossRef] [PubMed]
- Timme, R.E.; Kuehl, J.V.; Boore, J.L.; Jansen, R.K. A comparative analysis of the Lactuca and Helianthus (Asteraceae) plastid genomes: Identification of divergent regions and categorization of shared repeats. Am. J. Bot. 2007, 94, 302–312. [Google Scholar] [CrossRef] [PubMed]
- Morton, B.R. The influence of neighboring base composition on substitutions in plant chloroplast coding sequences. Mol. Biol. Evol. 1997, 14, 189–194. [Google Scholar] [CrossRef]
- Yan, X.; Liu, T.; Yuan, X.; Xu, Y.; Yan, H.; Hao, G. Chloroplast genomes and comparative analyses among thirteen taxa within Myrsinaceae s. str. clade (Myrsinoideae, Primulaceae). Int. J. Mol. Sci. 2019, 20, 4534. [Google Scholar] [CrossRef] [PubMed]
- Nadeem, M.A.; Nawaz, M.A.; Shahid, M.Q.; Doğan, Y.; Comertpay, G.; Yıldız, M.; Hatipoğlu, R.; Ahmad, F.; Alsaleh, A.; Labhane, N.; et al. DNA molecular markers in plant breeding: Current status and recent advancements in genomic selection and genome editing. Biotechnol. Biotechnol. Equip. 2018, 32, 261–285. [Google Scholar] [CrossRef]
- Mohammad-Panah, N.; Shabanian, N.; Khadivi, A.; Rahmani, M.S.; Emami, A. Genetic structure of gall oak (Quercus infectoria) characterized by nuclear and chloroplast SSR markers. Tree Genet. Genomes 2017, 13, 70. [Google Scholar] [CrossRef]
- Xu, C.; Cai, X.; Chen, Q.; Zhou, H.; Cai, Y.; Ben, A. Factors affecting synonymous codon usage bias in chloroplast genome of oncidium gower ramsey. Evol. Bioinform. 2011, 7, 271–278. [Google Scholar] [CrossRef]
- Chakraborty, S.; Yengkhom, S.; Uddin, A. Analysis of codon usage bias of chloroplast genes in Oryza species: Codon usage of chloroplast genes in Oryza species. Planta 2020, 252, 67. [Google Scholar] [CrossRef]
- Yang, Y.; Zhu, J.; Feng, L.; Zhou, T.; Bai, G.; Yang, J.; Zhao, G. Plastid genome comparative and phylogenetic analyses of the key genera in Fagaceae: Highlighting the effect of codon composition bias in phylogenetic inference. Front. Plant Sci. 2018, 9, 82. [Google Scholar] [CrossRef]
- Chi, X.; Zhang, F.; Dong, Q.; Chen, S. Insights into Comparative Genomics, Codon Usage Bias, and Phylogenetic Relationship of Species from Biebersteiniaceae and Nitrariaceae Based on Complete Chloroplast Genomes. Plants 2020, 9, 1605. [Google Scholar] [CrossRef]
- Dong, W.; Liu, J.; Yu, J.; Wang, L.; Zhou, S. Highly variable chloroplast markers for evaluating plant phylogeny at low taxonomic levels and for DNA barcoding. PLoS ONE 2012, 7, e35071. [Google Scholar] [CrossRef] [PubMed]
- Dong, W.; Xu, C.; Li, C.; Sun, J.; Zuo, Y.; Shi, S.; Cheng, T.; Guo, J.; Zhou, S. Ycf1, the most promising plastid DNA barcode of land plants. Sci. Rep. 2015, 5, 8348. [Google Scholar] [CrossRef] [PubMed]
- Cavender-Bares, J.; González-Rodríguez, A.; Eaton, D.A.; Hipp, A.A.; Beulke, A.; Manos, P.S. Phylogeny and biogeography of the American live oaks (Quercus subsection Virentes): A genomic and population genetics approach. Mol. Ecol. 2015, 24, 3668–3687. [Google Scholar] [CrossRef] [PubMed]
- Eaton, D.A.; Hipp, A.L.; González-Rodríguez, A.; Cavender-Bares, J. Historical introgression among the American live oaks and the comparative nature of tests for introgression. Evolution 2015, 69, 2587–2601. [Google Scholar] [CrossRef] [PubMed]
- Gugger, P.F.; Cavender-Bares, J. Molecular and morphological support for a Florida origin of the Cuban oak. J. Biogeogr. 2013, 40, 632–645. [Google Scholar] [CrossRef]
- Hipp, A.L.; Eaton, D.A.; Cavender-Bares, J.; Fitzek, E.; Nipper, R.; Manos, P.S. A framework phylogeny of the American oak clade based on sequenced RAD data. PLoS ONE 2014, 9, e93975. [Google Scholar] [CrossRef]
- Manos, P.S.; Doyle, J.J.; Nixon, K.C. Phylogeny, biogeography, and processes of molecular differentiation in Quercus subgenus Quercus (Fagaceae). Mol. Phylogenetics Evol. 1999, 12, 333–349. [Google Scholar] [CrossRef]
- Petit, R.J.; Bodénès, C.; Ducousso, A.; Roussel, G.; Kremer, A. Hybridization as a mechanism of invasion in oaks. New Phytol. 2004, 161, 151–164. [Google Scholar] [CrossRef]
- Burgarella, C.; Lorenzo, Z.; Jabbour-Zahab, R.; Lumaret, R.; Guichoux, E.; Petit, R.J.; Soto, A.; Gil, L. Detection of hybrids in nature: Application to oaks (Quercus suber and Q. ilex). Heredity 2009, 102, 442–452. [Google Scholar] [CrossRef] [PubMed]
- Leroy, T.; Louvet, J.M.; Lalanne, C.; Le Provost, G.; Labadie, K.; Aury, J.M.; Delzon, S.; Plomion, C.; Kremer, A. Adaptive introgression as a driver of local adaptation to climate in European white oaks. New Phytol. 2020, 226, 1171–1182. [Google Scholar] [CrossRef] [PubMed]
- Ortego, J.; Gugger, P.F.; Riordan, E.C.; Sork, V.L. Influence of climatic niche suitability and geographical overlap on hybridization patterns among southern Californian oaks. J. Biogeogr. 2014, 41, 1895–1908. [Google Scholar] [CrossRef]
- Rushton, B.S. Natural hybridization within the genus Quercus L. Ann. For. Sci. 1993, 50, 73s–90s. [Google Scholar] [CrossRef]
- Hipp, A.L.; Manos, P.S.; Hahn, M.; Avishai, M.; Bodénès, C.; Cavender-Bares, J.; Crowl, A.A.; Deng, M.; Denk, T.; Fitz-Gibbon, S.; et al. Genomic landscape of the global oak phylogeny. New Phytol. 2020, 226, 1198–1212. [Google Scholar] [CrossRef] [PubMed]
- Hudson, G.S.; Mason, J.G. The chloroplast genes encoding subunits of the H+-ATP synthase. Photosynth. Res. 1988, 18, 205–222. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, P. Chemiosmotic coupling in oxidative and photosynthetic phosphorylation. Biol. Rev. Camb. Philos. Soc. 1966, 41, 445–502. [Google Scholar] [CrossRef] [PubMed]
- Martin, M.; Casano, L.M.; Sabater, B. Identification of the product of ndhA gene as a thylakoid protein synthesized in response to photooxidative treatment. Plant Cell Physiol. 1996, 37, 293–298. [Google Scholar] [CrossRef] [PubMed]
- Endo, T.; Shikanai, T.; Takabayashi, A.; Asada, K.; Sato, F. The role of chloroplastic NAD(P)H dehydrogenase in photoprotection. FEBS Lett. 1999, 457, 5–8. [Google Scholar] [CrossRef] [PubMed]
- Martin, M.; Sabater, B. Plastid ndh genes in plant evolution. Plant Physiol. Biochem. 2010, 48, 636–645. [Google Scholar] [CrossRef] [PubMed]
- Martin-Avila, E.; Lim, Y.-L.; Birch, R.; Dirk, L.M.; Buck, S.; Rhodes, T.; Sharwood, R.E.; Kapralov, M.V.; Whitney, S.M. Modifying plant photosynthesis and growth via simultaneous chloroplast transformation of Rubisco large and small subunits. Plant Cell 2020, 32, 2898–2916. [Google Scholar] [CrossRef]
- Chen, X.; Kindle, K.; Stern, D. Initiation codon mutations in the Chlamydomonas chloroplast petD gene result in temperature-sensitive photosynthetic growth. EMBO J. 1993, 12, 3627–3635. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Voucher No. | GenBank Accession No. | Latitude (N) | Longitude (E) | Place of Collection |
|---|---|---|---|---|---|
| Q. poilanei | DM15650 | OR835153 | 23.416667 | 108.36667 | Daming Mountain, China |
| Q. helferiana | DM19757 | OR835154 | 18.495611 | 99.302050 | Kun Tan National Park, Thailand |
| Q. camusiae | DM19880 | OR966887 | 18.539589 | 98.534078 | Mae Klang Luang Trail, Thailand |
| Q. semiserrata | DM19890 | OR966888 | 18.541483 | 98.543278 | Mae Klang Luang Trail, Thailand |
| Species | Q. poilanei | Q. helferiana | Q. camusiae | Q. semiserrata |
|---|---|---|---|---|
| Genome size (bp) | 160,784 | 160,878 | 161,632 | 161,630 |
| Length of LSC (bp) | 90,216 | 90,343 | 90,294 | 90,292 |
| Length of IRs (a/b) (bp) | 25,842 | 25,829 | 26,593 | 26,593 |
| Length of SSC (bp) | 18,884 | 18,877 | 18,152 | 18,152 |
| Total GC content (%) | 36.9 | 36.9 | 36.9 | 36.9 |
| GC content of LSC (%) | 34.74 | 34.74 | 34.75 | 34.75 |
| GC content of IRs (%) | 42.77 | 42.70 | 42.35 | 42.35 |
| GC content of SSC (%) | 31.11 | 31.11 | 31.22 | 31.22 |
| Number of genes | 131 | 131 | 131 | 131 |
| Number of PCGs | 86 | 86 | 86 | 86 |
| Number of tRNAs | 37 | 37 | 37 | 37 |
| Number of rRNAs | 8 | 8 | 8 | 8 |
| Category | Gene Group | Gene Name |
|---|---|---|
| Photosynthesis | Photosystem I | psaA, psaB, psaC, psaI, psaJ |
| Photosystem II | psbA, psbB, psbC, psbD, psbE, psbF, psbH, psbI, psbJ, psbK, psbL, psbM, psbN, psbT, psbZ | |
| NADH dehydrogenase | ndhA*, ndhB*(×2), ndhC, ndhD, ndhE, ndhF, ndhG, ndhH, ndhI, ndhJ, ndhK | |
| Cytochrome b/f complex | petA, petB*, petD*, petG, petL, petN | |
| ATP synthase | atpA, atpB, atpE, atpF*, atpH, atpI | |
| Rubisco of Large subunit | rbcL | |
| Transcription and translation | Translation initiation factor | infA |
| Ribosomal Proteins (LSU) | rpl14, rpl16*, rpl2*(×2), rpl20, rpl22, rpl23(×2), rpl32, rpl33, rpl36 | |
| Ribosomal Proteins (SSU) | rps11, rps12**(×2), rps14, rps15, rps16*, rps18, rps19, rps2, rps3, rps4, rps7(×2), rps8 | |
| RNA polymerase | rpoA, rpoB, rpoC1*, rpoC2 | |
| Ribosomal RNAs | rrn16(×2), rrn23(×2), rrn4.5(×2), rrn5(×2) | |
| Transfer RNAs | trnA-UGC*(×2), trnC-GCA, trnD-GUC, trnE-UUC, trnF-GAA, trnG-GCC, trnG-UCC*, trnH-GUG, trnI-CAU(×2), trnI-GAU*(×2), trnK-UUU*, trnL-CAA(×2), trnL-UAA*, trnL-UAG, trnM-CAU, trnN-GUU(×2), trnP-UGG, trnQ-UUG, trnR-ACG(×2), trnR-UCU, trnS-GCU, trnS-GGA, trnS-UGA, trnT-GGU, trnT-UGU, trnV-GAC(×2), trnV-UAC*, trnW-CCA, trnY-GUA, trnfM-CAU | |
| Biosynthesis | Maturase | matK |
| ATP-dependendent Protease | clpP1** | |
| Acetyl-CoA carboxylase | accD | |
| Envelope membrane protein | cemA | |
| C-type cytochrome synthesis gene | ccsA | |
| Unknown | Conserved hypothetical chloroplast ORF | ycf1(×2), ycf2(×2), ycf3 **, ycf4 |
| Species | No. (Proportion) of SSRs | Distribution of SSRs | |||||
|---|---|---|---|---|---|---|---|
| LSC | SSC | IRs | IGS | CDS | Intron | ||
| Q. poilanei | 116 (24.32%) | 88 | 18 | 10 | 84 | 16 | 16 |
| Q. helferiana | 115 (24.10%) | 87 | 20 | 8 | 80 | 16 | 19 |
| Q. camusiae | 123 (25.79%) | 90 | 23 | 10 | 85 | 20 | 18 |
| Q. semiserrata | 123 (25.79%) | 90 | 23 | 10 | 85 | 20 | 18 |
| Total | 477 (100%) | 355 (74.4%) | 84 (17.6%) | 38 (8%) | 334 (70%) | 72 (15.1%) | 71 (14.9%) |
| Species | No. of Repeat Sequences | Length Distribution of M | Length Distribution of D | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| M | F | R | P | C | 9 | 10–19 | 20–29 | 31 | 30 | 31–40 | 41–50 | 51–60 | 64 | |
| Q. poilanei | 31 | 14 | 3 | 21 | 1 | 0 | 18 | 11 | 2 | 15 | 22 | 1 | 1 | 0 |
| Q. helferiana | 30 | 18 | 2 | 23 | 0 | 1 | 19 | 8 | 2 | 20 | 17 | 5 | 1 | 0 |
| Q. camusiae | 28 | 15 | 2 | 19 | 0 | 1 | 19 | 7 | 2 | 15 | 19 | 1 | 0 | 1 |
| Q. semiserrata | 28 | 15 | 2 | 19 | 0 | 0 | 19 | 7 | 2 | 16 | 18 | 1 | 0 | 1 |
| Total | 117 | 62 | 9 | 82 | 1 | 2 | 75 | 33 | 8 | 66 | 76 | 8 | 2 | 2 |
| Gene | atpF | ||
|---|---|---|---|
| Model Comparison | M0 vs. M3 | M1 vs. M2 | M7 vs. M8 |
| df | 4 | 2 | 2 |
| ΔlnL | 36.484478 | 21.965641 | 22.022258 |
| 2ΔlnL | 72.968956 | 43.931282 | 44.044516 |
| LRT (p-value) | 5.35604 × 10−15 | 2.88698 × 10−10 | 2.72807 × 10−10 |
| Positively selected sites | / | 17A (0.621), 49S (0.996 **), 50D (0.993 **), 52N (0.994 **), 54R (1.000 **), 104N (0.545) | 17A (0.674), 49S (0.998 **), 50D (0.997 **), 52N (0.998 **), 54R (1.000 **), 104N (0.598) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, L.-L.; Li, Y.; Zheng, S.-S.; Kozlowski, G.; Xu, J.; Song, Y.-G. Complete Chloroplast Genomes of Four Oaks from the Section Cyclobalanopsis Improve the Phylogenetic Analysis and Understanding of Evolutionary Processes in the Genus Quercus. Genes 2024, 15, 230. https://doi.org/10.3390/genes15020230
Wang L-L, Li Y, Zheng S-S, Kozlowski G, Xu J, Song Y-G. Complete Chloroplast Genomes of Four Oaks from the Section Cyclobalanopsis Improve the Phylogenetic Analysis and Understanding of Evolutionary Processes in the Genus Quercus. Genes. 2024; 15(2):230. https://doi.org/10.3390/genes15020230
Chicago/Turabian StyleWang, Ling-Ling, Yu Li, Si-Si Zheng, Gregor Kozlowski, Jin Xu, and Yi-Gang Song. 2024. "Complete Chloroplast Genomes of Four Oaks from the Section Cyclobalanopsis Improve the Phylogenetic Analysis and Understanding of Evolutionary Processes in the Genus Quercus" Genes 15, no. 2: 230. https://doi.org/10.3390/genes15020230