
Comparative Analysis of the Complete Chloroplast Genomes in Allium Section Bromatorrhiza Species (Amaryllidaceae): Phylogenetic Relationship and Adaptive Evolution


Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Sample Collection, DNA Extraction, and Complete Genome Sequencing
2.2. Chloroplast Genome Sequence Assembly and Annotation
2.3. Contraction and Expansion of IRs and Repeat Content
2.4. Codon Usage Bias Analysis and RNA Editing Sites
2.5. Sequence Divergence and Nucleotide Diversity Analysis
2.6. Phylogenetic Tree Analysis
2.7. Positive Selection Analysis
3. Results
3.1. Genome Features of sect. Bromatorrhiza
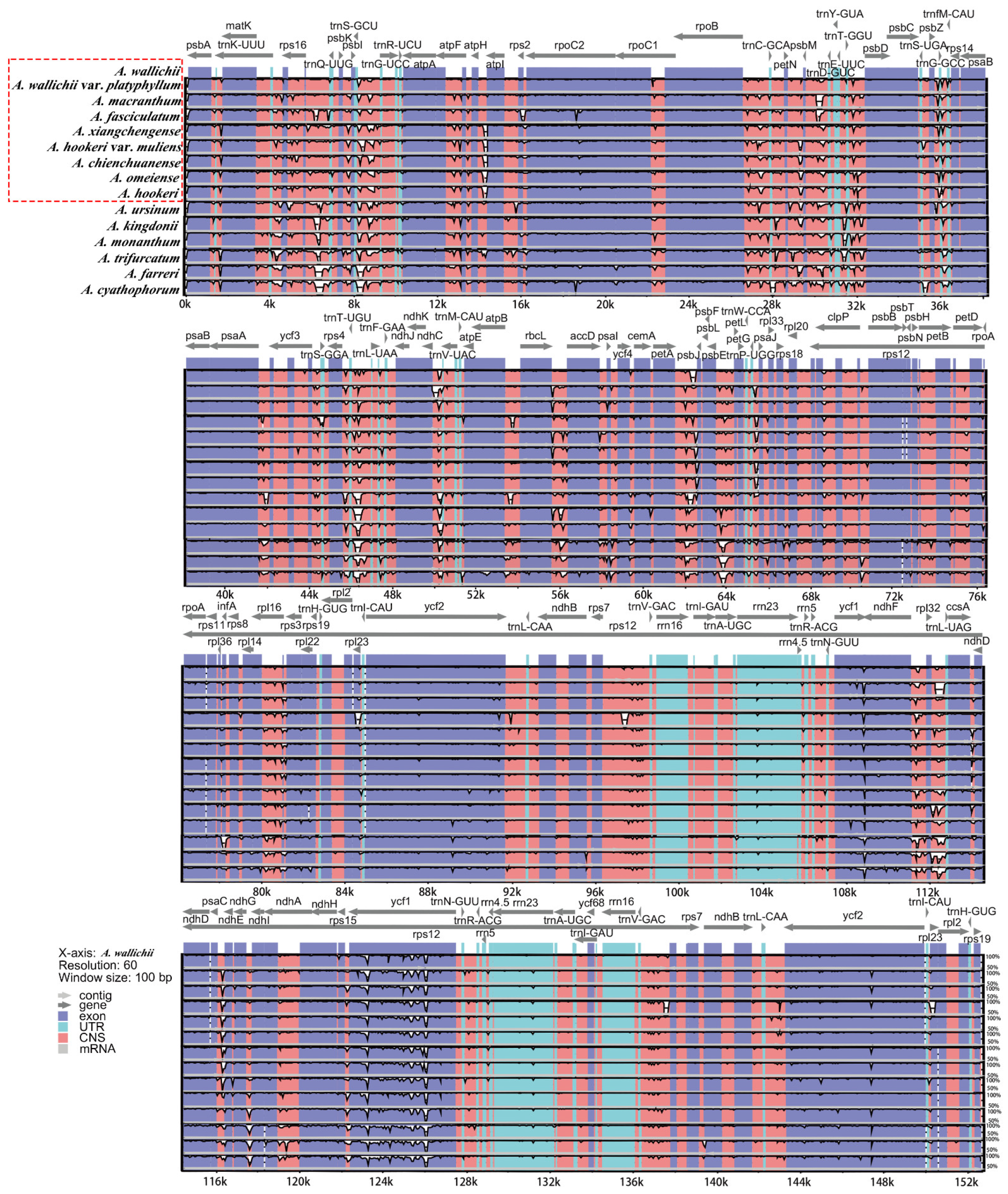
3.2. Comparative Analysis of the Chloroplast Genome Structure of sect. Bromatorrhiza
3.3. Codon Usage Bias and RNA Editing Site Analysis
3.4. Sequence Divergence Analysis
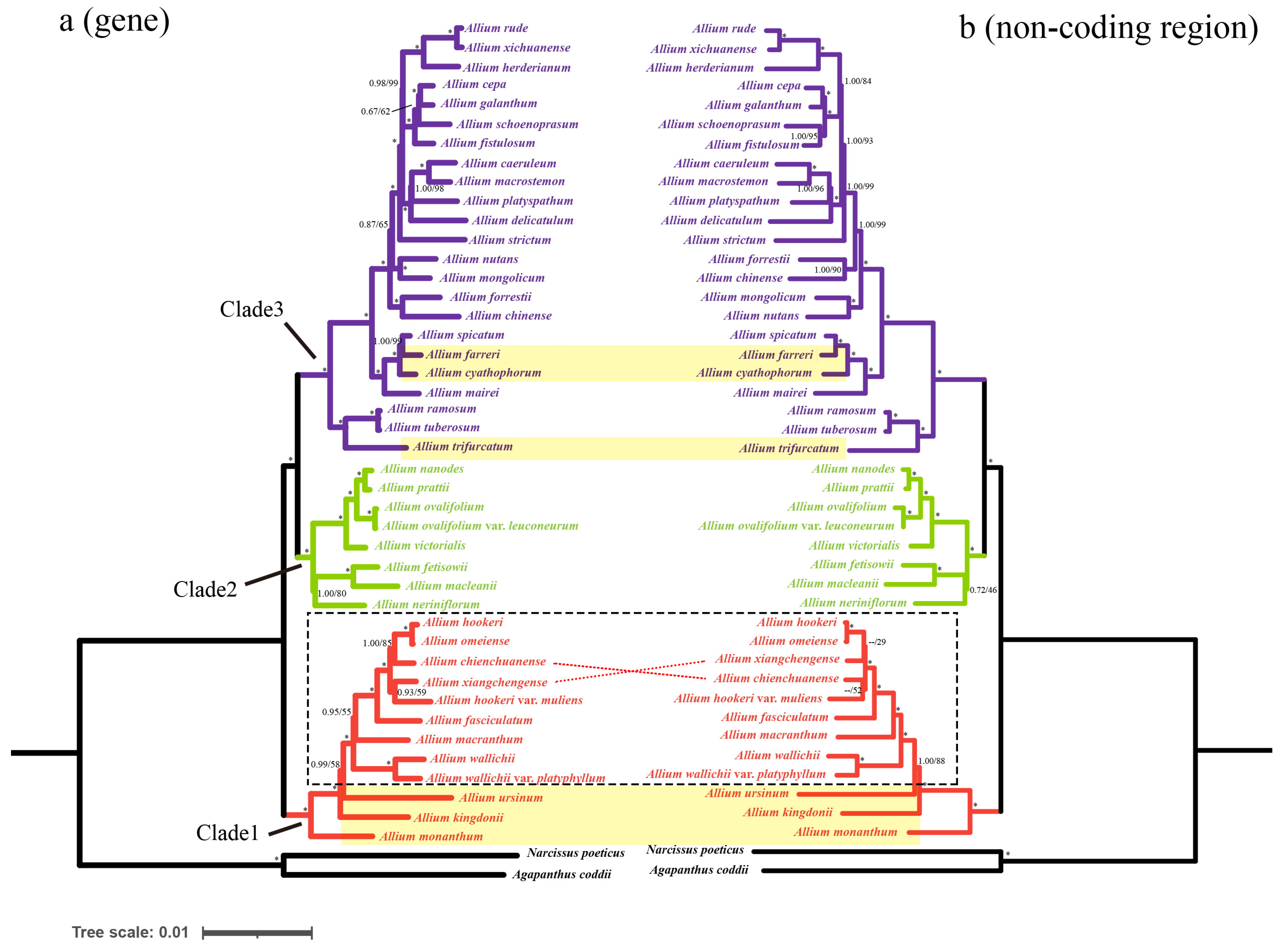
3.5. Phylogenetic Analysis
3.6. Adaptive Evolution Analysis
4. Discussion
4.1. Complete Chloroplast Genome Structure
4.2. cp Genome Sequence Variation and Potential DNA Barcode Markers
4.3. Phylogenetic Analysis
4.4. Selective Pressure Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Govaerts, R.; Kington, S.; Friesen, N.; Fritsch, R.; Snijman, D.A.; Marcucci, R.; Silverstone-Sopkin, P.A.B.S. World checklist of Amaryllidaceae. Facilitated by the Royal Botanic Gardens, Kew 2005–2020. Available online: http://wcsp.science.kew.org/ (accessed on 21 October 2021).
- Fritsch, R.M.; Friesen, N. Evolution, domestication and taxonomy. In Allium Crop Science: Recent Advances; Rabinowitch, H.D., Currah, L., Eds.; CABI Publishing: Wallingford, UK, 2002; pp. 5–30. [Google Scholar]
- Xu, J.M.; Kamelin, R.V.; Allium, L. Flora of China; Wu, Z.Y., Raven, P.H., Eds.; Science Press: Beijing, China; Missouri Botanical Garden Press: St. Louis, MO, USA, 2000; Volume 24, pp. 165–202. [Google Scholar]
- Aryal, K.P.; Poudel, S.; Chaudhary, R.P.; Chettri, N.; Chaudhary, P.; Ning, W.; Kotru, R. Diversity and use of wild and non-cultivated edible plants in the Western Himalaya. J. Ethnobiol. Ethnomed. 2018, 14, 10. [Google Scholar] [CrossRef] [PubMed]
- Bhandari, J.; Muhammad, B.; Thapa, P.; Shrestha, B.G. Study of phytochemical, anti-microbial, anti-oxidant, and anti-cancer properties of Allium wallichii. BMC Complement. Altern. Med. 2017, 17, 102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ebrahimi, R.; Zamani, Z.; Kashi, A. Genetic diversity evaluation of wild Persian shallot (Allium hirtifolium Boiss.) using morphological and RAPD markers. Sci. Hortic. 2009, 119, 345–351. [Google Scholar] [CrossRef]
- Friesen, N.; Fritsch, R.M.; Blattner, F.R. Phylogeny and New Intrageneric Classification of Allium (Alliaceae) Based on Nuclear Ribosomal DNA ITS Sequences. Aliso 2006, 22, 372–395. [Google Scholar] [CrossRef]
- Fritsch, R.M.; Blattner, F.R.; Gurushidze, M. New classification of Allium, L. subg. Melanocrommyum (Webb & Berthel.) Rouy (Alliaceae) based on molecular and morphological characters. Phyton Ann. Rei Bot. 2010, 49, 145–320. [Google Scholar]
- Wheeler, E.J.; Mashayekhi, S.; McNeal, D.W.; Columbus, J.T.; Pires, J.C. Molecular systematics of Allium subgenus Amerallium (Amaryllidaceae) in North America. Am. J. Bot. 2013, 100, 701–711. [Google Scholar] [CrossRef]
- Li, Q.Q.; Zhou, S.D.; He, X.J.; Yu, Y.; Zhang, Y.C.; Wei, X.Q. Phylogeny and biogeography of Allium (Amaryllidaceae: Allieae) based on nuclear ribosomal internal transcribed spacer and chloroplast rps16 sequences, focusing on the inclusion of species endemic to China. Ann. Bot. 2010, 106, 709–733. [Google Scholar] [CrossRef]
- Huang, D.Q.; Yang, J.T.; Zhou, C.J.; Zhou, S.D.; He, X.J. Phylogenetic reappraisal of Allium subgenus Cyathophora (Amaryllidaceae) and related taxa, with a proposal of two new sections. J. Plant Res. 2014, 127, 275–286. [Google Scholar] [CrossRef]
- Li, Q.Q.; Zhou, S.D.; Huang, D.Q.; He, X.J.; Wei, X.Q. Molecular phylogeny, divergence time estimates and historical biogeography within one of the world’s largest monocot genera. AoB Plants 2016, 8, plw041. [Google Scholar] [CrossRef] [Green Version]
- Li, M.J.; Tan, J.B.; Xie, D.F.; Huang, D.Q.; Gao, Y.D.; He, X.J. Revisiting the evolutionary events in Allium subgenus Cyathophora (Amaryllidaceae): Insights into the effect of the Hengduan Mountains Region (HMR) uplift and Quaternary climatic fluctuations to the environmental changes in the Qinghai-Tibet Plateau. Mol. Phylogenet. Evol. 2016, 94, 802–813. [Google Scholar] [CrossRef]
- Xu, J.M.; Allium, L. Flora Reipublicae Popularis Sinicae; Wang, F.T., Tang, J., Eds.; Science Press: Beijing, China, 1980; Volume 14, pp. 170–272. [Google Scholar]
- Ekberg, L. Studies in the genus Allium II: A new subgenus and new sections from Asia. Bot. Not. 1969, 122, 57–68. [Google Scholar]
- Hanelt, P.; Schultze-Motel, J.; Fritsch, R.; Kruse, J.; Maab, H.I.; Ohle, H.; Pistrick, K. Infrageneric grouping of Allium—the Gatersleben approach. In The Genus Allium—Taxonomic Problems and Genetic Resources; Hanelt, P., Hammer, K., Knupffer, H., Eds.; IPK: Gatersleben, Germany, 1992; pp. 107–123. [Google Scholar]
- Hanelt, P.; Reinhard, F. Notes on Some Infrageneric Taxa in Allium, L. Kew Bull. 1994, 49, 559–564. [Google Scholar] [CrossRef]
- Samoylov, A.; Klaas, M.; Hanelt, P. Use of chloroplast DNA polymorphisms for the phylogenetic study of the subgenera Amerallium and Bromatorrhiza (genus Allium). Feddes Repert. 1995, 106, 161–167. [Google Scholar] [CrossRef]
- Samoylov, A.; Friesen, N.; Pollner, S.; Hanelt, P. Use of chloroplast DNA polymorphisms for the phylogenetic study of Allium subgenus Amerallium and subgenus Bromatorrhiza (Alliaceae) II. Feddes Repert. 1999, 110, 103–109. [Google Scholar] [CrossRef]
- Xue, P.F. Study on Karyotypes and their Relationships in Allium sect. Bromatorrhiza from China. Master’s Thesis, Sichuan University, Chengdu, China, 1994. [Google Scholar]
- Huang, R.F.; Xu, J.M.; Yu, H. A study on karyotypes and their evolutionary trends in Allium sect. Bromatorrhiza Ekberg (Liliaceae). Cathaya 1995, 7, 133–145. [Google Scholar]
- Klaas, M.; Friesen, N.; Rabinowitch, H.D.; Currah, L. 8 Molecular Markers in Allium; UKCABI: Wallingford, UK, 2002. [Google Scholar]
- Li, Q.Q.; Zhou, S.D.; He, X.J.; Wei, X.Q. Phylogeny and Character Evolution in Allium Subgenus Amerallium (Amaryllidaceae). Plant Divers. Resour. 2012, 34, 107–119. [Google Scholar] [CrossRef]
- Li, M.J.; Guo, X.L.; Li, J.; Zhou, S.D.; He, X.J. Cytotaxonomy of Allium (Amaryllidaceae) subgenera Cyathophora and Amerallium sect. Bromatorrhiza. Phytotaxa 2017, 331, 185. [Google Scholar] [CrossRef]
- Li, M.J.; Liu, J.Q.; Guo, X.L.; Xiao, Q.Y.; He, X.J. Taxonomic revision of Allium cyathophorum (Amaryllidaceae). Phytotaxa 2019, 415, 240–246. [Google Scholar] [CrossRef]
- Huang, D.Q. Molecular Systematics of Allium subgenus Cyathophora, Sections Bromatorrhiza and Sikkimensia (Amaryllidaceae) and Intraspecific Genetic Differentiation of A. wallichii. Ph.D. Thesis, Sichuan University, Chengdu, China, 2014. [Google Scholar]
- Sharma, G.; Gohil, R.N.; Kaul, V. Cytological status of Allium hookeri Thwaites (2n = 22). Genet. Resour. Crop Evol. 2011, 58, 1041–1050. [Google Scholar] [CrossRef]
- Douglas, S.E. Chloroplast origins and evolution. In The Molecular Biology of Cyanobacteria; Bryant, D.A., Ed.; Springer: Dordrecht, The Netherlands, 1994; pp. 91–118. [Google Scholar]
- Raubeson, L.A.; Jansen, R.K.; Henry, R.J. Chloroplast genomes of plants. In Plant Diversity and Evolution: Genotypic and Phenotypic Variation in Higher Plants; Henry, R.J., Ed.; CABI Press: Cambridge, MA, USA, 2005; pp. 45–68. [Google Scholar]
- Wolfe, K.H.; Li, W.H.; Sharp, P.M. Rates of nucleotide substitution vary greatly among plant mitochondrial, chloroplast, and nuclear DNAs. Proc. Natl. Acad. Sci. USA 1987, 84, 9054–9058. [Google Scholar] [CrossRef] [Green Version]
- Parks, M.; Cronn, R.; Liston, A. Increasing phylogenetic resolution at low taxonomic levels using massively parallel sequencing of chloroplast genomes. BMC Biol. 2009, 7, 84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wen, J.; Xie, D.F.; Price, M.; Ren, T.; Deng, Y.Q.; Gui, L.J.; Guo, X.L.; He, X.J. Backbone phylogeny and evolution of Apioideae (Apiaceae): New insights from phylogenomic analyses of plastome data. Mol. Phylogenet. Evol. 2021, 161, 107183. [Google Scholar] [CrossRef]
- Liu, C.K.; Lei, J.Q.; Jiang, Q.P.; Zhou, S.D.; He, X.J. The complete plastomes of seven Peucedanum plants: Comparative and phylogenetic analyses for the Peucedanum genus. BMC Plant Biol. 2022, 22, 101. [Google Scholar] [CrossRef] [PubMed]
- Dong, W.P.; Xu, C.; Li, C.H.; Sun, J.H.; Zuo, Y.J.; Shi, S.; Cheng, T.; Guo, J.J.; Zhou, S.L. ycf1, the most promising plastid DNA barcode of land plants. Sci. Rep. 2015, 5, 8348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.; Chon, J.K.; Lim, J.S.; Kim, E.K.; Nah, G. Characterization of Complete Chloroplast Genome of Allium victorialis and Its Application for Barcode Markers. Plant Breed. Biotechnol. 2017, 5, 221–227. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, V.B.; Park, H.S.; Lee, S.C.; Lee, J.; Park, J.Y.; Yang, T.J. Authentication Markers for Five Major Panax Species Developed via Comparative Analysis of Complete Chloroplast Genome Sequences. J. Agric. Food Chem. 2017, 65, 6298–6306. [Google Scholar] [CrossRef] [PubMed]
- Xie, D.F.; Tan, J.B.; Yu, Y.; Gui, L.J.; Su, D.M.; Zhou, S.D.; He, X.J. Insights into phylogeny, age and evolution of Allium (Amaryllidaceae) based on the whole plastome sequences. Ann. Bot. 2020, 125, 1039–1055. [Google Scholar] [CrossRef]
- Ren, T.; Li, Z.X.; Xie, D.F.; Gui, L.J.; Peng, C.; Wen, J.; He, X.J. Plastomes of eight Ligusticum species: Characterization, genome evolution, and phylogenetic relationships. BMC Plant Biol. 2020, 20, 519. [Google Scholar] [CrossRef]
- Xie, D.F.; Yu, H.X.; Price, M.; Xie, C.; Deng, Y.Q.; Chen, J.P.; Yu, Y.; Zhou, S.D.; He, X.J. Phylogeny of Chinese Allium Species in Section Daghestanica and Adaptive Evolution of Allium (Amaryllidaceae, Allioideae) Species Revealed by the Chloroplast Complete Genome. Front. Plant Sci. 2019, 10, 460. [Google Scholar] [CrossRef] [Green Version]
- Omelchenko, D.O.; Krinitsina, A.A.; Belenikin, M.S.; Konorov, E.A.; Kuptsov, S.V.; Logacheva, M.D.; Speranskaya, A.S. Complete plastome sequencing of Allium paradoxum reveals unusual rearrangements and the loss of the ndh genes as compared to Allium ursinum and other onions. Gene 2020, 726, 144154. [Google Scholar] [CrossRef]
- Li, H.; Xie, D.F.; Chen, J.P.; Zhou, S.D.; He, X.J. Chloroplast genomic comparison of two sister species Allium macranthum and A. fasciculatum provides valuable insights into adaptive evolution. Genes Genom. 2020, 42, 507–517. [Google Scholar] [CrossRef]
- Yusupov, Z.; Deng, T.; Volis, S.; Khassanov, F.; Makhmudjanov, D.; Tojibaev, K.; Sun, H. Phylogenomics of Allium section Cepa (Amaryllidaceae) provides new insights on domestication of onion. Plant Divers. 2021, 43, 102–110. [Google Scholar] [CrossRef] [PubMed]
- Namgung, J.; Do, H.D.K.; Kim, C.; Choi, H.J.; Kim, J.H. Complete chloroplast genomes shed light on phylogenetic relationships, divergence time, and biogeography of Allioideae (Amaryllidaceae). Sci. Rep. 2021, 11, 3262. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Xie, D.F.; Chen, J.P.; Zhou, S.D.; Yu, Y.; He, X.J. Comparative Analysis of the Complete Chloroplast Genomes in Allium Subgenus Cyathophora (Amaryllidaceae): Phylogenetic Relationship and Adaptive Evolution. BioMed Res. Int. 2020, 2020, 1732586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dierckxsens, N.; Mardulyn, P.; Smits, G. NOVOPlasty: De novo assembly of organelle genomes from whole genome data. Nucleic Acids Res. 2017, 45, e18. [Google Scholar]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef]
- Qu, X.J.; Moore, M.J.; Li, D.Z.; Yi, T.S. PGA: A software package for rapid, accurate, and flexible batch annotation of plastomes. Plant Methods 2019, 15, 50. [Google Scholar] [CrossRef] [Green Version]
- Lowe, T.M.; Chan, P.P. tRNAscan-SE On-line: Integrating search and context for analysis of transfer RNA genes. Nucleic Acids Res. 2016, 44, W54–W57. [Google Scholar] [CrossRef]
- Greiner, S.; Lehwark, P.; Bock, R. OrganellarGenomeDRAW (OGDRAW) version 1.3.1: Expanded toolkit for the graphical visualization of organellar genomes. Nucleic Acids Res. 2019, 47, W59–W64. [Google Scholar] [CrossRef] [Green Version]
- Amiryousefi, A.; Hyvönen, J.; Poczai, P. IRscope: An online program to visualize the junction sites of chloroplast genomes. Bioinformatics 2018, 34, 3030–3031. [Google Scholar] [CrossRef]
- Thiel, T.; Michalek, W.; Varshney, R.; Graner, A. Exploiting EST databases for the development and characterization of gene-derived SSR-markers in barley (Hordeum vulgare L.). Theor. Appl. Genet. 2003, 106, 411–422. [Google Scholar] [CrossRef] [PubMed]
- Kurtz, S.; Choudhuri, J.V.; Ohlebusch, E.; Schleiermacher, C.; Stoye, J.; Giegerich, R. REPuter: The manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 2001, 29, 4633–4642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharp, P.M.; Li, W.-H. An evolutionary perspective on synonymous codon usage in unicellular organisms. J. Mol. Evol. 1986, 24, 28–38. [Google Scholar] [CrossRef]
- Wright, F. The effective number of codons’ used in a gene. Gene 1990, 87, 23–29. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular Evolutionary Genetics Analysis Version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mower, J.P. The PREP suite: Predictive RNA editors for plant mitochondrial genes, chloroplast genes and user-defined alignments. Nucleic Acids Res. 2009, 37 (Suppl. 2), W253–W259. [Google Scholar] [CrossRef]
- Frazer, K.A.; Pachter, L.; Poliakov, A.; Rubin, E.M.; Dubchak, I. VISTA: Computational tools for comparative genomics. Nucleic Acids Res. 2004, 32 (Suppl. 2), W273–W279. [Google Scholar] [CrossRef]
- Darling, A.C.E.; Mau, B.; Blattner, F.R.; Perna, N.T. Mauve: Multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 2004, 14, 1394–1403. [Google Scholar] [CrossRef] [Green Version]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Librado, P.; Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009, 25, 1451–1452. [Google Scholar] [CrossRef] [Green Version]
- Posada, D.; Crandall, K.A. MODELTEST: Testing the model of DNA substitution. Bioinformatics 1998, 14, 817–818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [PubMed]
- Ronquist, F.; Teslenko, M.; van der Mark, P.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, D.P.; Zhang, Y.B.; Zhang, Z.; Zhu, J.; Yu, J. KaKs_Calculator 2.0: A toolkit incorporating γ-series methods and sliding window strategies. Genom. Proteom. Bioinform. 2010, 8, 77–80. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.H.; Nielsen, R. Codon-Substitution Models for Detecting Molecular Adaptation at Individual Sites Along Specific Lineages. Mol. Biol. Evol. 2002, 19, 908–917. [Google Scholar] [CrossRef] [Green Version]
- Lan, Y.; Sun, J.; Tian, R.; Bartlett, D.H.; Li, R.; Wong, Y.H.; Zhang, W.; Qiu, J.-W.; Xu, T.; He, L.-S.; et al. Molecular adaptation in the world’s deepest-living animal: Insights from transcriptome sequencing of the hadal amphipod Hirondellea gigas. Mol. Ecol. 2017, 26, 3732–3743. [Google Scholar] [CrossRef]
- Reis, M.d.; Yang, Z. Approximate Likelihood Calculation on a Phylogeny for Bayesian Estimation of Divergence Times. Mol. Biol. Evol. 2011, 28, 2161–2172. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.H.; Wong, W.S.W.; Nielsen, R. Bayes Empirical Bayes Inference of Amino Acid Sites Under Positive Selection. Mol. Biol. Evol. 2005, 22, 1107–1118. [Google Scholar] [CrossRef] [Green Version]
- Huo, Y.M.; Gao, L.M.; Liu, B.J.; Yang, Y.Y.; Kong, S.P.; Sun, Y.Q.; Yang, Y.H.; Wu, X. Complete chloroplast genome sequences of four Allium species: Comparative and phylogenetic analyses. Sci. Rep. 2019, 9, 12250. [Google Scholar] [CrossRef] [Green Version]
- Guo, X.L.; Zheng, H.Y.; Price, M.; Zhou, S.D.; He, X.J. Phylogeny and Comparative Analysis of Chinese Chamaesium Species Revealed by the Complete Plastid Genome. Plants 2020, 9, 965. [Google Scholar] [CrossRef]
- Filyushin, M.A.; Beletsky, A.V.; Mazur, A.M.; Kochieva, E.Z. The complete plastid genome sequence of garlic Allium sativum L. Mitochondrial DNA Part B 2016, 1, 831–832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wakasugi, T.; Tsudzuki, J.; Ito, S.; Nakashima, K.; Tsudzuki, T.; Sugiura, M. Loss of All ndh Genes as Determined by Sequencing the Entire Chloroplast Genome of the Black Pine Pinus thunbergii. Proc. Natl. Acad. Sci. USA 1994, 91, 9794–9798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, J.B.; Xia, H.A.; Cao, M.L.; Zhang, X.Q.; Zeng, W.Y.; Hu, S.N.; Tong, W.; Wang, J.; Wang, J.; Yu, J.; et al. A Comparison of Rice Chloroplast Genomes. Plant Physiol. 2004, 135, 412–420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wicke, S.; Naumann, J. Chapter eleven—Molecular evolution of plastid genomes in parasitic flowering plants. In Advances in Botanical Research; Chaw, S.-M., Jansen, R.K., Eds.; Academic Press: Cambridge, MA, USA, 2018; Volume 85, pp. 315–347. [Google Scholar]
- Samigullin, T.H.; Logacheva, M.D.; Terenteva, E.I.; Degtjareva, G.V.; Vallejo-Roman, C.M. Plastid genome of Seseli montanum: Complete sequence and comparison with plastomes of other members of the Apiaceae family. Biochemistry 2016, 81, 981–985. [Google Scholar] [CrossRef] [PubMed]
- Kelchner, S.A. The Evolution of Non-Coding Chloroplast DNA and Its Application in Plant Systematics. Ann. Mo. Bot. Gard. 2000, 87, 482–498. [Google Scholar] [CrossRef]
- Yin, K.Q.; Zhang, Y.; Li, Y.J.; Du, F.K. Different Natural Selection Pressures on the atpF Gene in Evergreen Sclerophyllous and Deciduous Oak Species: Evidence from Comparative Analysis of the Complete Chloroplast Genome of Quercus aquifolioides with Other Oak Species. Int. J. Mol. Sci. 2018, 19, 1042. [Google Scholar] [CrossRef] [Green Version]
- Naydenov, K.D.; Naydenov, M.K.; Alexandrov, A.; Vasilevski, K.; Gyuleva, V.; Matevski, V.; Nikolic, B.; Goudiaby, V.; Bogunic, F.; Paitaridou, D.; et al. Ancient split of major genetic lineages of European Black Pine: Evidence from chloroplast DNA. Tree Genet. Genomes 2016, 12, 68. [Google Scholar] [CrossRef]
- Zhang, X.; Zhou, T.; Yang, J.; Sun, J.J.; Ju, M.M.; Zhao, Y.M.; Zhao, G.F. Comparative Analyses of Chloroplast Genomes of Cucurbitaceae Species: Lights into Selective Pressures and Phylogenetic Relationships. Molecules 2018, 23, 2165. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Price, M.; Su, D.M.; Zhang, Z.; Yu, Y.; Xie, D.F.; Zhou, S.D.; He, X.J.; Gao, X.F. Phylogeny and Comparative Analysis for the Plastid Genomes of Five Tulipa (Liliaceae). BioMed Res. Int. 2021, 2021, 6648429. [Google Scholar] [CrossRef]
- Li, J.; Xie, D.F.; Guo, X.L.; Zheng, Z.Y.; He, X.J.; Zhou, S.D. Comparative Analysis of the Complete Plastid Genome of Five Bupleurum Species and New Insights into DNA Barcoding and Phylogenetic Relationship. Plants 2020, 9, 543. [Google Scholar] [CrossRef]
- Huang, L.S.; Sun, Y.Q.; Jin, Y.Q.; Gao, Q.; Hu, X.G.; Gao, F.L.; Yang, X.L.; Zhu, J.J.; El-Kassaby, Y.A.; Mao, J.F. Development of high transferability cpSSR markers for individual identification and genetic investigation in Cupressaceae species. Ecol. Evol. 2018, 8, 4967–4977. [Google Scholar] [CrossRef] [Green Version]
- Bull, L.N.; Pabón-Peña, C.R.; Freimer, N.B. Compound microsatellite repeats: Practical and theoretical features. Genome Res. 1999, 9, 830–838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khakhlova, O.; Bock, R. Elimination of deleterious mutations in plastid genomes by gene conversion. Plant J. 2006, 46, 85–94. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.C.; Zhou, T.; Duan, D.; Yang, J.; Feng, L.; Zhao, G.F. Comparative Analysis of the Complete Chloroplast Genomes of Five Quercus Species. Front. Plant Sci. 2016, 7, 959. [Google Scholar] [CrossRef] [Green Version]
- Li, X.W.; Yang, Y.; Henry, R.J.; Rossetto, M.; Wang, Y.T.; Chen, S.L. Plant DNA barcoding: From gene to genome. Biol. Rev. 2015, 90, 157–166. [Google Scholar] [CrossRef]
- Dong, W.L.; Wang, R.N.; Zhang, N.Y.; Fan, W.B.; Fang, M.F.; Li, Z.H. Molecular Evolution of Chloroplast Genomes of Orchid Species: Insights into Phylogenetic Relationship and Adaptive Evolution. Int. J. Mol. Sci. 2018, 19, 716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, G.; Wei, L.L.; Ma, L.; Wu, Z.Q.; Gu, C.H.; Chen, K. Comparative analyses of chloroplast genomes from 13 Lagerstroemia (Lythraceae) species: Identification of highly divergent regions and inference of phylogenetic relationships. Plant Mol. Biol. 2020, 102, 659–676. [Google Scholar] [CrossRef]
- Henriquez, C.L.; Abdullah; Ahmed, I.; Carlsen, M.M.; Zuluaga, A.; Croat, T.B.; McKain, M.R. Evolutionary dynamics of chloroplast genomes in subfamily Aroideae (Araceae). Genomics 2020, 112, 2349–2360. [Google Scholar] [CrossRef]
- Huang, R.; Xie, X.N.; Chen, A.M.; Li, F.; Tian, E.W.; Chao, Z. The chloroplast genomes of four Bupleurum (Apiaceae) species endemic to Southwestern China, a diversity center of the genus, as well as their evolutionary implications and phylogenetic inferences. BMC Genom. 2021, 22, 714. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, N.H.; Driscoll, H.E.; Specht, C.D. A molecular phylogeny of the wild onions (Allium; Alliaceae) with a focus on the western North American center of diversity. Mol. Phylogenet. Evol. 2008, 47, 1157–1172. [Google Scholar] [CrossRef]
- Hanaoka, M.; Kato, M.; Anma, M.; Tanaka, K. SIG1, a sigma factor for the chloroplast RNA polymerase, differently associates with multiple DNA regions in the chloroplast chromosomes in vivo. Int. J. Mol. Sci. 2012, 13, 12182–12194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, S.L.; Sablok, G.; Wang, B.; Qu, D.; Barbaro, E.; Viola, R.; Li, M.G.; Varotto, C. Plastome organization and evolution of chloroplast genes in Cardamine species adapted to contrasting habitats. BMC Genom. 2015, 16, 306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Macadlo, L.A.; Ibrahim, I.M.; Puthiyaveetil, S. Sigma factor 1 in chloroplast gene transcription and photosynthetic light acclimation. J. Exp. Bot. 2019, 71, 1029–1038. [Google Scholar] [CrossRef] [PubMed]
- Hargreaves, A.L.; Samis, K.E.; Eckert, C.G. Are species’ range limits simply niche limits writ large? A review of transplant experiments beyond the range. Am. Nat. 2014, 183, 157–173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raven, J.A.; Beardall, J.; Larkum, A.W.D.; Sánchez-Baracaldo, P. Interactions of photosynthesis with genome size and function. Philos. Trans. R. Soc. B Biol. Sci. 2013, 368, 20120264. [Google Scholar] [CrossRef]
- Scobeyeva, V.A.; Artyushin, I.V.; Krinitsina, A.A.; Nikitin, P.A.; Antipin, M.I.; Kuptsov, S.V.; Belenikin, M.S.; Omelchenko, D.O.; Logacheva, M.D.; Konorov, E.A.; et al. Gene Loss, Pseudogenization in Plastomes of Genus Allium (Amaryllidaceae), and Putative Selection for Adaptation to Environmental Conditions. Front. Genet. 2021, 12, 674783. [Google Scholar] [CrossRef]
- Gao, X.Y.; Zhang, X.; Meng, H.H.; Li, J.; Zhang, D.; Liu, C.N. Comparative chloroplast genomes of Paris Sect. Marmorata: Insights into repeat regions and evolutionary implications. BMC Genom. 2018, 19, 878. [Google Scholar] [CrossRef]
- Kode, V.; Mudd, E.A.; Iamtham, S.; Day, A. The tobacco plastid accD gene is essential and is required for leaf development. Plant J. 2005, 44, 237–244. [Google Scholar] [CrossRef]
- Wicke, S.; Schneeweiss, G.M.; dePamphilis, C.W.; Müller, K.F.; Quandt, D. The evolution of the plastid chromosome in land plants: Gene content, gene order, gene function. Plant Mol. Biol. 2011, 76, 273–297. [Google Scholar] [CrossRef] [Green Version]
- Sugita, M.; Shinozaki, K.; Sugiura, M. Tobacco Chloroplast tRNALys(UUU) Gene Contains a 2.5-Kilobasepair Intron: An Open Reading Frame and a Conserved Boundary Sequence in the Intron. Proc. Natl. Acad. Sci. USA 1985, 82, 3557–3561. [Google Scholar] [CrossRef] [Green Version]
- Ems, S.C.; Morden, C.W.; Dixon, C.K.; Wolfe, K.H.; de Pamphilis, C.W.; Palmer, J.D. Transcription, splicing and editing of plastid RNAs in the nonphotosynthetic plant Epifagus virginiana. Plant Mol. Biol. 1995, 29, 721–733. [Google Scholar] [CrossRef] [PubMed]
- Martín, M.; Casano, L.M.; Sabater, B. Identification of the product of ndhA gene as a thylakoid protein synthesized in response to photooxidative treatment. Plant Cell Physiol. 1996, 37, 293–298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bent, A.F.; Kunkel, B.N.; Dahlbeck, D.; Brown, K.L.; Schmidt, R.; Giraudat, J.; Leung, J.; Staskawicz, B.J. RPS2 of Arabidopsis thaliana: A Leucine-Rich Repeat Class of Plant Disease Resistance Genes. Science 1994, 265, 1856–1860. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, S.; Bédard, J.; Hirano, M.; Hirabayashi, Y.; Oishi, M.; Imai, M.; Takase, M.; Ide, T.; Nakai, M. Uncovering the Protein Translocon at the Chloroplast Inner Envelope Membrane. Science 2013, 339, 571–574. [Google Scholar] [CrossRef]
- Zhong, Q.W.; Yang, S.P.; Sun, X.M.; Wang, L.H.; Li, Y. The complete chloroplast genome of the Jerusalem artichoke (Helianthus tuberosus L.) and an adaptive evolutionary analysis of the ycf2 gene. PeerJ 2019, 7, e7596. [Google Scholar] [CrossRef] [Green Version]
- Xie, F.M.; Xie, D.F.; Xie, C.; Yu, Y.; Zhou, S.D.; He, X.J. Adaptation Evolution and Phylogenetic Analyses of Species in Chinese Allium Section Pallasia and Related Species Based on Complete Chloroplast Genome Sequences. BioMed Res. Int. 2020, 2020, 8542797. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | rbcL Accession | NCBI Accession | Length(bp) | GC Contents (%) | Number of Genes | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Genome | LSC | SSC | IR | Genome | LSC | SSC | IR | Total | PCG | rRNA | tRNA | |||
| A. wallichii | KP207707 | ON184003 | 152615 | 82283 | 17138 | 26597 | 37.0 | 34.9 | 30.3 | 42.5 | 134 | 88 | 8 | 38 |
| A. wallichii var. platyphyllum | JX017625 | ON184004 | 152926 | 82557 | 17121 | 26624 | 37.0 | 34.8 | 30.2 | 42.5 | 134 | 88 | 8 | 38 |
| A. macranthum | JX017626 | ON184005 | 152596 | 82168 | 17606 | 26411 | 37.1 | 34.9 | 30.4 | 42.7 | 134 | 88 | 8 | 38 |
| A. fasciculatum | JX017623 | ON184006 | 153214 | 82240 | 17406 | 26784 | 37.0 | 34.9 | 30.1 | 42.6 | 134 | 88 | 8 | 38 |
| A. xiangchengense | JX017621 | ON184007 | 152294 | 82378 | 17404 | 26256 | 37.0 | 34.8 | 30.1 | 42.7 | 134 | 88 | 8 | 38 |
| A. hookeri var. muliens | JX017621 | ON184008 | 153113 | 82366 | 17361 | 26693 | 37.0 | 34.8 | 30.1 | 42.6 | 134 | 88 | 8 | 38 |
| A. chienchuanense | JX017621 | ON184009 | 153549 | 82542 | 17357 | 26825 | 37.0 | 34.8 | 30.1 | 42.5 | 134 | 88 | 8 | 38 |
| A. omeiense | JX017621 | ON184010 | 153592 | 82609 | 17387 | 26798 | 37.0 | 34.8 | 30.0 | 42.6 | 134 | 88 | 8 | 38 |
| A. hookeri | JX017621 | ON184011 | 153682 | 82700 | 17386 | 26798 | 37.0 | 34.8 | 30.0 | 42.6 | 134 | 88 | 8 | 38 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, J.; Xie, D.; He, X.; Yang, Y.; Li, X. Comparative Analysis of the Complete Chloroplast Genomes in Allium Section Bromatorrhiza Species (Amaryllidaceae): Phylogenetic Relationship and Adaptive Evolution. Genes 2022, 13, 1279. https://doi.org/10.3390/genes13071279
Chen J, Xie D, He X, Yang Y, Li X. Comparative Analysis of the Complete Chloroplast Genomes in Allium Section Bromatorrhiza Species (Amaryllidaceae): Phylogenetic Relationship and Adaptive Evolution. Genes. 2022; 13(7):1279. https://doi.org/10.3390/genes13071279
Chicago/Turabian StyleChen, Junpei, Dengfeng Xie, Xingjin He, Yi Yang, and Xufeng Li. 2022. "Comparative Analysis of the Complete Chloroplast Genomes in Allium Section Bromatorrhiza Species (Amaryllidaceae): Phylogenetic Relationship and Adaptive Evolution" Genes 13, no. 7: 1279. https://doi.org/10.3390/genes13071279