
Comparative Chloroplast Genomics of 21 Species in Zingiberales with Implications for Their Phylogenetic Relationships and Molecular Dating


Abstract
:1. Introduction
2. Results
2.1. Characteristics of 22 Complete Chloroplast Genomes in Zingiberales
2.2. Repeat Sequences Analyses
2.3. Codons Usage Analysis
2.4. Comparative Analysis of 22 Complete Chloroplast Genomes in Zingiberales
2.5. Highly Divergence Regions and Selective Pressure Analyses of Zingiberales
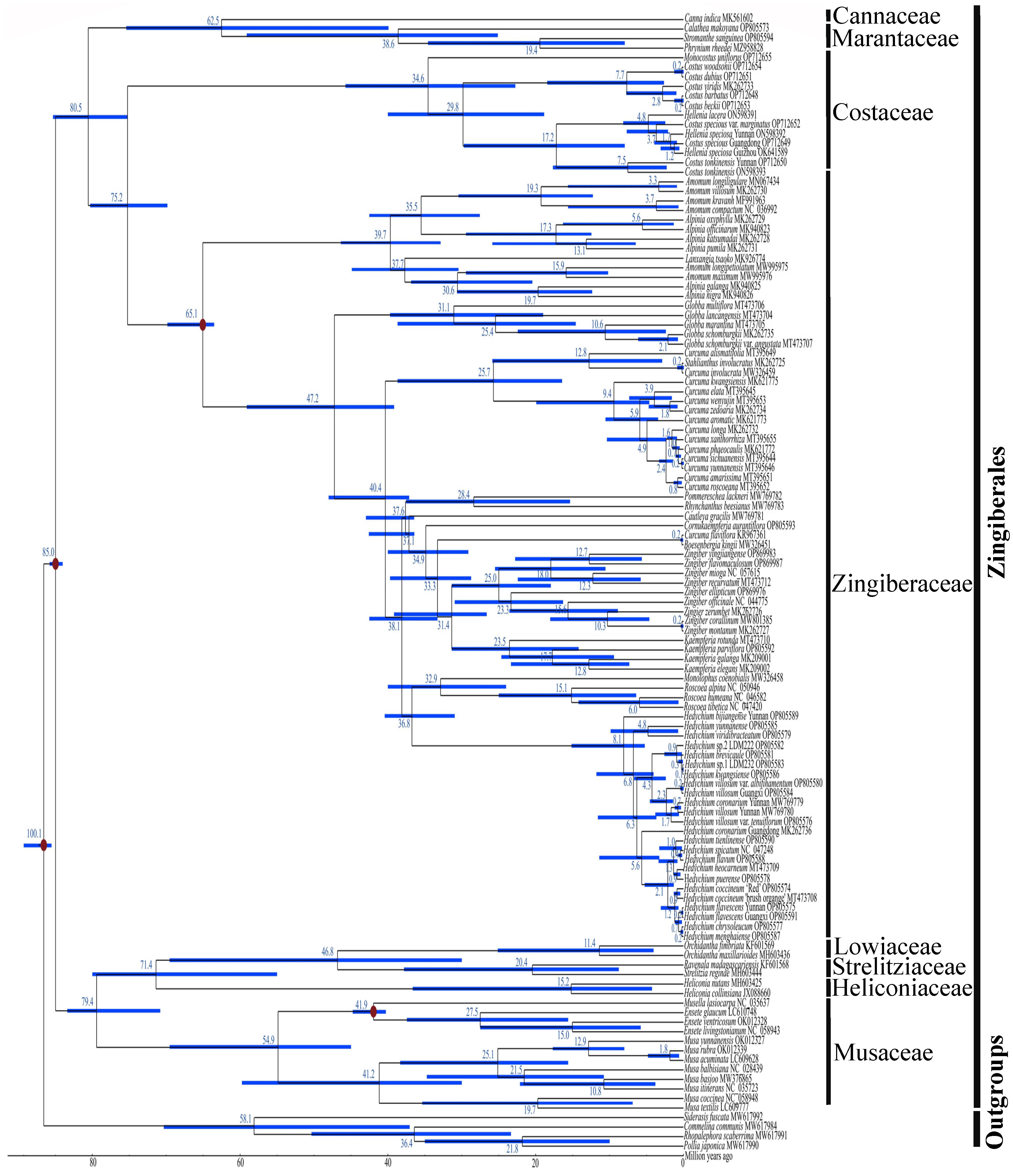
2.6. Phylogenetic Relationships of Zingiberales
2.7. Divergence Time Estimation of Zingiberales
3. Discussion
3.1. Genome Structure Comparison and Sequence Variation
3.2. Positive Selection and Phylogenetic Analysis within Zingiberales
3.3. Divergence Time within Zingiberales
4. Materials and Methods
4.1. Plant Sample Collection, DNA Extraction, and Sequencing
4.2. Chloroplast Genome Assembly and Annotation
4.3. Repeat Sequences Analysis
4.4. Codon Usage Analysis
4.5. Chloroplast Genomes Comparison
4.6. Nucleotide Diversity and Gene Selective Pressure Analyses
4.7. Phylogenetic Relationship Analysis
4.8. Divergence Time Estimation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| bp | Base paIRs |
| BLAST | Basic Local Alignment Search Tool |
| NCBI | National Center for Biotechnology Information |
| IR | Inverted repeat |
| ITS | Internal transcribed spacer |
| LSC | Large single copy |
| SSC | Small single copy |
| SSRs | Simple sequence repeats |
| Mya | Million years ago |
| ML | Maximum likelihood |
| BI | Bayesian inference |
| HPD | Highest posterior density |
| G | Guanine |
| C | Cytosine |
| DNA | Deoxyribonucleic acid |
| RNA | Ribonucleic acid |
| tRNA | Transfer RNA |
| rRNA | Ribosomal RNA |
References
- Kress, W.J. The phylogeny and classification of the Zingiberales. Ann. Missouri. Bot. Gard. 1990, 77, 698–721. [Google Scholar] [CrossRef]
- Kress, W.J.; Prince, L.M.; Hahn, W.J.; Zimmer, E.A. Unraveling the evolutionary radiation of the families of the Zingiberales using morphological and molecular evidence. Syst. Biol. 2001, 50, 926–944. [Google Scholar] [CrossRef] [PubMed]
- Barrett, C.F.; Specht, C.D.; Leebens-Mack, J.; Stevenson, D.W.; Zomlefer, W.B.; Davis, J.I. Resolving ancient radiations: Can complete plastid gene sets elucidate deep relationships among the tropical gingers (Zingiberales)? Ann. Bot. 2014, 113, 119–133. [Google Scholar] [CrossRef]
- Sass, C.; Iles, W.J.D.; Barrett, C.F.; Smith, S.Y.; Specht, C.D. Revisiting the Zingiberales: Using multiplexed exon capture to resolve ancient and recent phylogenetic splits in a charismatic plant lineage. Peer J. 2016, 4, e1584. [Google Scholar] [CrossRef] [PubMed]
- Carlsen, M.M.; Fér, T.; Schmickl, R.; Leong-Škorničková, J.; Newman, M.; Kress, W.J. Resolving the rapid plant radiation of early diverging lineages in the tropical Zingiberales: Pushing the limits of genomic data. Mol. Phylogenet. Evol. 2018, 128, 55–68. [Google Scholar] [CrossRef] [PubMed]
- Givnish, T.J.; Zuluaga, A.; Spalink, D.; Soto Gomez, M.; Lam, V.K.Y.; Saarela, J.M.; Sass, C.; Iles, W.J.D.; de Sousa, D.J.L.; Leebens-Mack, J.; et al. Monocot plastid phylogenomics, timeline, net rates of species diversification, the power of multi-gene analyses, and a functional model for the origin of monocots. Am. J. Bot. 2018, 105, 1888–1910. [Google Scholar] [CrossRef] [PubMed]
- Kress, W.J.; Specht, C.D. The evolutionary and biogeographic origin and diversification of the tropical monocot order Zingiberales. Aliso 2006, 22, 621–632. [Google Scholar] [CrossRef]
- Smith, S.Y.; Iles, W.J.D.; Benedict, J.C.; Specht, C.D. Building the monocot tree of death: Progress and challenges emerging from the macrofossil-rich Zingiberales. Am. J. Bot. 2018, 105, 1389–1400. [Google Scholar] [CrossRef]
- Fu, N.; Ji, M.; Rouard, M.; Yan, H.F.; Ge, X.J. Comparative chloroplast genome analysis of Musaceae and new insights into phylogenetic relationships. BMC Genom. 2022, 23, 223. [Google Scholar] [CrossRef]
- Cui, Y.; Nie, L.; Sun, W.; Xu, Z.; Wang, Y.; Yu, J.; Song, J.; Yao, H. Comparative and phylogenetic analyses of ginger (Zingiber officinale) in the family Zingiberaceae based on the complete chloroplast genome. Plants 2019, 8, 283. [Google Scholar] [CrossRef]
- Cui, Y.; Chen, X.; Nie, L.; Sun, W.; Hu, H.; Lin, Y.; Li, H.; Zheng, X.; Song, J.; Yao, H. Comparison and phylogenetic analysis of chloroplast genomes of three medicinal and edible Amomum species. Int. J. Mol. Sci. 2019, 20, 4040. [Google Scholar] [CrossRef] [PubMed]
- Li, D.M.; Zhao, C.Y.; Zhu, G.F.; Xu, Y.C. Complete chloroplast genome sequence of Hedychium coronarium. Mitochondrial DNA B. 2019, 4, 2806–2807. [Google Scholar] [CrossRef] [PubMed]
- Li, D.M.; Zhu, G.F.; Xu, Y.C.; Ye, Y.J.; Liu, J.M. Complete chloroplast genomes of three medicinal Alpinia species: Genome organization, comparative analyses and phylogenetic relationships in family Zingiberaceae. Plants 2020, 9, 286. [Google Scholar] [CrossRef] [PubMed]
- Liang, H.; Zhang, Y.; Deng, J.; Gao, G.; Ding, C.; Zhang, L.; Yang, R. The complete chloroplast genome sequences of 14 Curcuma species: Insights into genome evolution and phylogenetic relationships within Zingiberales. Front. Genet. 2020, 11, 802. [Google Scholar] [CrossRef]
- Yang, Q.; Fu, G.F.; Wu, Z.Q.; Li, L.; Zhao, J.L.; Li, Q.J. Chloroplast genome evolution in four montane Zingiberaceae taxa in China. Front. Plant Sci. 2022, 12, 774482. [Google Scholar] [CrossRef]
- André, T.; Salzman, S.; Wendt, T.; Specht, C.D. Speciation dynamics and biogeography of Neotropical spIRal gingers (Costaceae). Mol. Phylogenet. Evol. 2016, 103, 55–63. [Google Scholar] [CrossRef]
- Valderrama, E.; Sass, C.; Pinilla-Vargas, M.; Skinner, D.; Maas, P.J.M.; Maas-van de Kamer, H.; Landis, J.B.; Guan, C.J.; Specht, C.D. Unraveling the spIRaling radiation: A phylogenomic analysis of Neotropical Costus L. Front. Plant Sci. 2020, 11, 1195. [Google Scholar] [CrossRef]
- Soltis, E.D.; Soltis, P.S. Contributions of plant molecular systematics to studies of molecular evolution. Plant Mol. Biol. 2000, 42, 45–75. [Google Scholar] [CrossRef]
- Li, D.M.; Li, J.; Wang, D.R.; Xu, Y.C.; Zhu, G.F. Molecular evolution of chloroplast genomes in subfamily Zingiberoideae (Zingiberaceae). BMC Plant Biol. 2021, 21, 558. [Google Scholar] [CrossRef]
- Timilsena, P.R.; Wafula, E.K.; Barrett, C.F.; Ayyampalayam, S.; McNeal, J.R.; Rentsch, J.D.; McKain, M.R.; Heyduk, K.; Harkess, A.; Villegente, M.; et al. Phylogenomic resolution of order- and family-level monocot relationships using 602 single-copy nuclear genes and 1375 BUSCO genes. Front. Plant Sci. 2022, 13, 876779. [Google Scholar] [CrossRef]
- Kress, W.J.; Prince, L.M.; Williams, K.J. The phylogeny and a new classification of the gingers (Zingiberaceae) evidence from molecular data. Am. J. Bot. 2002, 89, 1682–1696. [Google Scholar] [CrossRef] [PubMed]
- Specht, C.D. Gondwanan vicariance or dispersal in the tropics? The biogeographic history of the tropical monocot family Costaceae (Zingiberales). Aliso 2006, 22, 631–642. [Google Scholar] [CrossRef]
- Johansen, L.B. Phylogeny of Orchidantha (Lowiaceae) and the Zingiberales based on six DNA regions. Syst. Biol. 2005, 30, 106–117. [Google Scholar] [CrossRef]
- Wood, T.H.; Whitten, W.M.; Williams, N.H. Phylogeny of Hedychium and related genera (Zingiberaceae) based on ITS sequence data. Edinb. J. Bot. 2000, 57, 261–270. [Google Scholar] [CrossRef]
- Techaprasan, J.; Klinbunga, S.; Ngamriabsakul, C.; Jenjittikul, T. Genetic variation of Kaempferia (Zingiberaceae) in Thailand based on chloroplast DNA (psbA-trnH and petA-psbJ) sequences. Genet Mol. Res. 2010, 9, 1957–1973. [Google Scholar] [CrossRef]
- Ashokan, A.; Xavier, A.; Suksathan, P.; Ardiyani, M.; Leong-Škorničková, J.; Newman, M.; Kress, W.J.; Gowda, V. Himalayan orogeny and monsoon intensification explain species diversification in an endemic ginger (Hedychium: Zingiberaceae) from the Indo-Malayan Realm. Mol. Phylogenet. Evol. 2022, 170, 107440. [Google Scholar] [CrossRef]
- Greiner, S.; Lehwark, P.; Bock, R. OrganellarGenomeDRAW (OGDRAW) version 1.3.1: Expanded toolkit for the graphical visualization of organellar genomes. Nucleic Acids Res. 2019, 47, W59–W64. [Google Scholar] [CrossRef]
- Grant, J.R.; Stothard, P. The CGView Server: A comparative genomics tool for circular genomes. Nucleic Acids Res. 2008, 36, W181–W184. [Google Scholar] [CrossRef]
- Frazer, K.A.; Pachter, L.; Poliakov, A.; Rubin, E.M.; Dubchak, I. VISTA: Computational tools for comparative genomics. Nucleic Acids Res. 2004, 32, W273–W279. [Google Scholar] [CrossRef]
- Frailey, D.C.; Chaluvadi, S.R.; Vaughn, J.N.; Coatney, C.G.; Bennetzen, J.L. Gene loss and genome rearrangement in the plastids of five Hemiparasites in the family Orobanchaceae. BMC Plant Biol. 2018, 18, 30. [Google Scholar] [CrossRef]
- Dong, W.L.; Wang, R.N.; Zhang, N.Y.; Fan, W.B.; Fang, M.F.; Li, Z.H. Molecular evolution of chloroplast genomes of orchid species: Insights into phylogenetic relationship and adaptive evolution. Int. J. Mol. Sci. 2018, 19, 716. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.F.; Landis, J.B.; Wang, H.X.; Zhu, Z.X.; Wang, H.F. Comparative analysis of chloroplast genome structure and molecular dating in Myrtales. BMC Plant Biol. 2021, 21, 219. [Google Scholar] [CrossRef] [PubMed]
- Henriquez, C.L.; Abdullah; Ahmed, I.; Carlsen, M.M.; Zuluaga, A.; Croat, T.B.; McKain, M.R. Molecular evolution of chloroplast genomes in Monsteroideae (Araceae). Planta 2020, 251, 72. [Google Scholar] [CrossRef]
- Kim, S.H.; Yang, J.; Park, J.; Yamada, T.; Maki, M.; Kim, S.C. Comparison of whole chloroplast genome sequences between thermogenic Skunk Cabbage Symplocarpus renifolius and Nonthermogenic S. nipponicus (Orontioideae; Araceae) in East Asia. Int. J. Mol. Sci. 2019, 20, 4678. [Google Scholar] [CrossRef]
- Gao, Y.; Liu, K.; Li, E.; Wang, Y.; Xu, C.; Zhao, L.; Dong, W. Dynamic evolution of the chloroplast genome in the Elm family (Ulmaceae). Planta 2022, 257, 14. [Google Scholar] [CrossRef] [PubMed]
- Zhou, D.; Mehmood, F.; Lin, P.; Cheng, T.; Wang, H.; Shi, S.; Zhang, J.; Meng, J.; Zheng, K.; Poczai, P. Characterization of the evolutionary pressure on Anisodus tanguticus Maxim. with complete chloroplast genome sequence. Genes 2022, 13, 2125. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Yang, N.; Dong, W.; Zhao, L. Molecular evolution and phylogenomic analysis of complete chloroplast genomes of Cotinus (Anacardiaceae). Ecol. Evol. 2023, 13, e10134. [Google Scholar] [CrossRef]
- Moghaddam, M.; Wojciechowski, M.F.; Kazempour-Osaloo, S. Characterization and comparative analysis of the complete plastid genomes of four Astragalus species. PLoS ONE 2023, 18, e0286083. [Google Scholar] [CrossRef]
- Yu, F.; Chen, Z.Y.; Liao, J.P.; Yu, H.P.; Wang, B.; Song, J.J.; Zou, P.; Ye, Y.S.; Huang, X.X.; Deng, Y.F.; et al. Botanical Paintings of Chinese Zingiberales; Huazong University of Science and Technology Press: Wuhan, China, 2012; Volume 1, pp. 162–169, 190–191. [Google Scholar]
- Wu, D.; Liu, N.; Ye, Y. The Zingiberaceous Resources in China; Huazhong University of Science and Technology University Press: Wuhan, China, 2016; Volume 1, pp. 109, 144, 165, 172. [Google Scholar]
- Li, X.; Hu, Z.; Lin, X.; Li, Q.; Gao, H.; Luo, G.; Chen, S. High-throughput pyrosequencing of the complete chloroplast genome of Magnolia officinalis and its application in species identification. Acta Pharm. Sin. 2012, 47, 124–130. [Google Scholar]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Jin, J.J.; Yu, W.B.; Yang, J.B.; Song, Y.; dePamphilis, C.W.; Yi, T.S.; Li, D.Z. GetOrganelle: A fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome Biol. 2020, 21, 241. [Google Scholar] [CrossRef]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef] [PubMed]
- Tillich, M.; Lehwark, P.; Pellizzer, T.; Ulbricht-Jones, E.S.; Fischer, A.; Bock, R.; Greiner, S. GeSeq—Versatile and accurate annotation of organelle genomes. Nucleic Acids Res. 2017, 45, W6–W11. [Google Scholar] [CrossRef] [PubMed]
- Wyman, S.K.; Jansen, R.K.; Boore, J.L. Automatic annotation of organellar genomes with DOGMA. Bioinformatics 2004, 20, 3252–3255. [Google Scholar] [CrossRef] [PubMed]
- Lowe, T.M.; Chan, P.P. tRNAscan-SE On-line: Search and contextual analysis of transfer RNA genes. Nucleic Acids Res. 2016, 44, W54–W57. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef]
- Lehwark, P.; Greiner, S. GB2sequin—A file converter preparing custom GenBank files for database submission. Genomics 2019, 111, 759–761. [Google Scholar] [CrossRef]
- Kurtz, S.; Choudhuri, J.V.; Ohlebusch, E.; Schleiermacher, C.; Stoye, J.; Giegerich, R. REPuter: The manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 2001, 29, 4633–4642. [Google Scholar] [CrossRef]
- Beier, S.; Thiel, T.; Münch, T.; Scholz, U.; Mascher, M. MISA-web: A web server for microsatellite prediction. Bioinformatics 2017, 33, 2583–2585. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Tamura, K. Mega 7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef]
- Amiryousefi, A.; Hyvönen, J.; Poczai, P. IRscope: An online program to visualize the junction sites of chloroplast genomes. Bioinformatics 2018, 34, 3030–3031. [Google Scholar] [CrossRef] [PubMed]
- Rozas, J.; Ferrer-Mata, A.; Sánchez-DelBarrio, J.C.; GuIRao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sánchez-Gracia, A. DnaSP 6: DNA sequence polymorphism analysis of large datasets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef] [PubMed]
- Gao, F.; Chen, C.; Arab, D.A.; Du, Z.; He, Y.; Ho, S.Y.W. EasyCodeML: A visual tool for analysis of selection using CodeML. Ecol. Evol. 2019, 9, 3891–3898. [Google Scholar] [CrossRef]
- Yang, Z. Likelihood ratio tests for detecting positive selection and application to primate lysozyme evolution. Mol. Biol. Evol. 1998, 15, 568–573. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Wong, W.S.W.; Nielsen, R. Bayes empirical bayes inference of amino acids sites under positive selection. Mol. Biol. Evol. 2005, 22, 1107–1118. [Google Scholar] [CrossRef] [PubMed]
- Rozewicki, J.; Li, S.; Amada, K.M.; Standley, D.M.; Katoh, K. MAFFT-DASH: Integrated protein sequence and structural alignment. Nucleic Acids Res. 2019, 47, W5–W10. [Google Scholar] [CrossRef]
- Santorum, J.M.; Darriba, D.; Taboada, G.L.; Posada, D. jmodeltest.org: Selection of nucleotide substitution models on the cloud. Bioinformatics 2014, 30, 1310–1311. [Google Scholar] [CrossRef]
- Guindon, S.; Dufayard, J.F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef]
- Ronquist, F.; Teslenko, M.; van der Mark, P.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef]
- Yang, Z. PAML 4: Phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 2007, 24, 1586–1591. [Google Scholar] [CrossRef]
- Hickey, L.J.; Peterson, R.K. Zingiberopsis, a fossil genus of the ginger family from the Late Cretaceous to Early Eocene sediments of western interior North America. Can. J. Bot. 1978, 56, 1136–1152. [Google Scholar] [CrossRef]
- Manchester, S.; Kress, W. Fossil banana (Musaceae): Ensete oregonense sp. nov. from the Eocene of western North America and its phytogeographic significance. Am. J. Bot. 1993, 80, 1264–1272. [Google Scholar] [CrossRef]
- Friis, E. Spirematospermum chandlerae sp. nov., an extinct species of Zingiberaceae from the North American Cretaceous. Tert. Res. 1988, 9, 7–12. [Google Scholar]
- Hertweck, K.L.; Kinney, M.S.; Stuart, S.A.; Maurin, O.; Mathews, S.; Chase, M.W.; Gandolfo, M.A.; Pires, J.C. Phylogenetics, divergence times and diversification from three genomic partitions in monocots. Bot. J. Linn. Soc. 2015, 178, 375–393. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species Name | GenBank Accession Number | Size (bp) | LSC (bp) | SSC (bp) | IR (bp) | GC Content (%) | Number of Genes (Different) | Number of CDS (Different) | Number of tRNA (Different) | Number of rRNA (Different) | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Total | LSC | SSC | IR | CDS | ||||||||||
| Hedychium bijiangense | OP805589 | 163,883 | 88,541 | 15,786 | 29,778 | 36.08 | 33.84 | 29.56 | 41.14 | 36.93 | 133 (112) | 87 (79) | 38 (29) | 8 (4) |
| Hedychium brevicaule | OP805581 | 163,438 | 88,016 | 15,862 | 29,780 | 36.09 | 33.86 | 29.52 | 41.14 | 36.91 | 131 (111) | 87 (79) | 36 (28) | 8 (4) |
| Hedychium chrysoleucum | OP805577 | 163,977 | 88,603 | 15,816 | 29,779 | 36.08 | 33.84 | 29.53 | 41.15 | 36.93 | 133 (112) | 87 (79) | 38 (29) | 8 (4) |
| Hedychium coccineum ‘Red’ | OP805574 | 163,850 | 88,487 | 15,805 | 29,779 | 36.10 | 33.88 | 29.54 | 41.15 | 36.93 | 133 (112) | 87 (79) | 38 (29) | 8 (4) |
| Hedychium flavescens Guangxi | OP805591 | 163,909 | 88,589 | 15,762 | 29,779 | 36.10 | 33.85 | 29.62 | 41.15 | 36.93 | 133 (112) | 87 (79) | 38 (29) | 8 (4) |
| Hedychium flavescens Yunnan | OP805575 | 163,951 | 88,591 | 15,804 | 29,778 | 36.09 | 33.85 | 29.57 | 41.15 | 36.93 | 133 (112) | 87 (79) | 38 (29) | 8 (4) |
| Hedychium flavum | OP805588 | 163,850 | 88,573 | 15,821 | 29,707 | 36.08 | 33.84 | 29.52 | 41.16 | 36.93 | 133 (112) | 87 (79) | 38 (29) | 8 (4) |
| Hedychium kwangsiense | OP805586 | 163,423 | 88,002 | 15,861 | 29,780 | 36.09 | 33.86 | 29.52 | 41.14 | 36.91 | 133 (112) | 87 (79) | 38 (29) | 8 (4) |
| Hedychium menghaiense | OP805587 | 163,979 | 88,603 | 15,818 | 29,779 | 36.08 | 33.84 | 29.53 | 41.15 | 36.93 | 133 (112) | 87 (79) | 38 (29) | 8 (4) |
| Hedychium puerense | OP805578 | 163,941 | 88,561 | 15,822 | 29,779 | 36.08 | 33.84 | 29.52 | 41.14 | 36.92 | 133 (112) | 87 (79) | 38 (29) | 8 (4) |
| Hedychium sp.1 LDM232 | OP805583 | 163,442 | 88,021 | 15,861 | 29,780 | 36.09 | 33.86 | 29.52 | 41.14 | 36.91 | 133 (112) | 87 (79) | 38 (29) | 8 (4) |
| Hedychium sp.2 LDM222 | OP805582 | 163,319 | 87,916 | 15,843 | 29,780 | 36.11 | 33.88 | 29.52 | 41.14 | 36.91 | 133 (112) | 87 (79) | 38 (29) | 8 (4) |
| Hedychium tienlinense | OP805590 | 163,868 | 88,488 | 15,820 | 29,780 | 36.09 | 33.87 | 29.53 | 41.14 | 36.93 | 133 (112) | 87 (79) | 38 (29) | 8 (4) |
| Hedychium villosum var. albifihamentum | OP805580 | 163,442 | 88,059 | 15,807 | 29,788 | 36.11 | 33.86 | 29.57 | 41.16 | 36.92 | 133 (112) | 87 (79) | 38 (29) | 8 (4) |
| Hedychium villosum Guangxi | OP805584 | 163,462 | 88,040 | 15,846 | 29,788 | 36.10 | 33.86 | 29.49 | 41.16 | 36.92 | 133 (112) | 87 (79) | 38 (29) | 8 (4) |
| Hedychium villosum var. tenuiflorum | OP805576 | 163,359 | 88,139 | 15,678 | 29,771 | 36.12 | 33.86 | 29.71 | 41.14 | 37.11 | 133 (112) | 87 (79) | 38 (29) | 8 (4) |
| Hedychium viridibracteatum | OP805579 | 163,338 | 88,032 | 15,746 | 29,780 | 36.12 | 33.89 | 29.61 | 41.15 | 36.93 | 133 (112) | 87 (79) | 38 (29) | 8 (4) |
| Hedychium yunnanense | OP805585 | 163,420 | 88,071 | 15,789 | 29,780 | 36.11 | 33.87 | 29.56 | 41.15 | 36.93 | 133 (112) | 87 (79) | 38 (29) | 8 (4) |
| Calathea makoyana | OP805573 | 166,906 | 91,910 | 20,848 | 27,074 | 36.67 | 34.93 | 30.35 | 42.05 | 37.60 | 134 (112) | 88 (79) | 38 (29) | 8 (4) |
| Cornukaempferia aurantiflora | OP805593 | 163,305 | 88,197 | 15,640 | 29,734 | 36.17 | 33.92 | 29.90 | 41.15 | 36.93 | 133 (112) | 87 (79) | 38 (29) | 8 (4) |
| Kaempferia parviflora | OP805592 | 163,075 | 87,769 | 15,812 | 29,747 | 36.16 | 33.97 | 29.57 | 41.14 | 36.95 | 133 (112) | 87 (79) | 38 (29) | 8 (4) |
| Stromanthe sanguinea | OP805594 | 161,303 | 87,056 | 18,937 | 27,655 | 36.55 | 34.44 | 29.84 | 42.16 | 37.57 | 133 (112) | 87 (79) | 38 (29) | 8 (4) |
| Category of Genes | Group of Genes | Name of Genes |
|---|---|---|
| Self-replication | DNA dependent RNA polymerase | rpoA, rpoB, rpoC1 *, rpoC2 |
| Large subunit of ribosomal proteins | rpl2 (×2) *, rpl14, rpl16 *, rpl20, rpl22 (×2) ①, rpl23 (×2), rpl32, rpl33, rpl36 | |
| Small subunit of ribosomal proteins | rps2, rps3, rps4, rps7 (×2), rps8, rps11, rps12 (×2) *, rps14, rps15, rps16 *, rps18, rps19 (×2) | |
| RNA genes | Ribosomal RNA | rrn4.5 (×2), rrn5 (×2), rrn16 (×2), rrn23 (×2) |
| Transfer RNA | trnA-UGC (×2) *, trnC-GCA, trnD-GUC, trnE-UUC, trnF-GAA, trnfM-CAU, trnG-GCC, trnG-UCC *, trnH-GUG (×2) ②, trnI-GAU (×2) *, trnK-UUU *, trnL-CAA (×2), trnL-UAA *, trnL-UAG, trnM-CAU (×3), trnN-GUU (×2), trnP-UGG, trnQ-UUG, trnR-ACG (×2), trnR-UCU ③, trnS-GCU, trnS-GGA, trnS-UGA, trnT-GGU, trnT-UGU, trnV-GAC (×2), trnV-UAC *, trnW-CCA, trnY-GUA | |
| Photosynthesis related genes | Subunits of photosystem Ⅰ | psaA, psaB, psaC, psaI, psaJ |
| Subunits of photosystem Ⅱ | psbA, psbB, psbC, psbD, psbE, psbF, psbH, psbI, psbJ, psbK, psbL, psbM, psbN, psbT, psbZ, infA | |
| Subunits of cytochrome b/f complex | petA, petB *, petD *, petG, petL, petN | |
| Subunits of ATP synthase | atpA, atpB, atpE, atpF *, atpH, atpI | |
| Subunits of NADH dehydrogenase | ndhA *, ndhB (×2) *, ndhC, ndhD, ndhE, ndhF, ndhG, ndhH, ndhI, ndhJ, ndhK | |
| Subunit of rubisco | rbcL | |
| Other genes | Subunit of acetyl-coA-carboxylase | accD |
| c-type cytochrome synthesis gene | ccsA | |
| Envelop membrane protein | cemA | |
| Protease | clpP ** | |
| Maturase | matK | |
| Genes of unknown function | Conserved open reading frames | ycf1 (×2), ycf2 (×2), ycf3 **, ycf4 |
| Regions | Length | Variable Sites | Informative Sites | Nucleotide Diversity | ||
|---|---|---|---|---|---|---|
| Number | % | Number | % | |||
| LSC | 88,541 | 65,855 | 74.38 | 54,324 | 61.35 | 0.1751 |
| SSC | 15,786 | 11,757 | 74.48 | 9131 | 57.84 | 0.1621 |
| IRa | 29,778 | 13,210 | 44.36 | 12,150 | 40.80 | 0.1455 |
| IRb | 29,778 | 20,298 | 68.16 | 17,141 | 57.56 | 0.1534 |
| Complete chloroplast genome | 163,883 | 111,120 | 67.80 | 92,746 | 56.59 | 0.1565 |
| Gene Names | Positively Selected Sites Pr (ω > 1) |
|---|---|
| atpA | 190 Q 0.981 *, 255 R 0.955 *, 459 V 0.998 ** |
| atpB | 83 M 0.975 *, 132 P 0.998 **, 143 L 0.996 **, 315 E 0.984 * |
| atpF | 49 L 0.959 *, 124 F 0.986 *, 178 A 0.966 * |
| atpI | 24 L 0.984 *, 65 D 0.990 * |
| clpP | 19 S 1.000 **, 21 V 0.999 **, 22 E 1.000 **, 38 S 0.996 **, 56 P 1.000 **, 73 S 0.968 *, 114 A 1.000 **, 128 G 0.989 *, 133 S 1.000 **, 136 A 0.985 *, 158 Y 0.957 *, 162 Y 0.982 *, 186 G 0.986 *, 187 I 0.966 *, 189 F 0.999 ** |
| matK | 92 V 0.955 *, 106 R 0.959 *, 146 W 0.985 *, 279 T 0.971 *, 294 F 0.985 *, 296 R 0.997 **, 413 P 0.970 * |
| ndhA | 132 F 0.982 * |
| ndhB | 13 F 0.996 **, 163 T 1.000 **, 228 P 0.993 **, 241 G 1.000 **, 379 S 1.000 ** |
| ndhC | 98 L 1.000 ** |
| ndhD | 422 A 0.989 * |
| ndhF | 504 G 0.998 **, 623 F 0.955 *, 630 K 0.957 * |
| ndhI | 165 D 0.967 * |
| petB | 1 M 1.000 **, 2 S 1.000 ** |
| petD | 106 T 0.990 ** |
| psaA | 152 S 0.968 *, 261 L 0.983 *, 292 A 0.988 * |
| psaI | 4 F 0.992 ** |
| psbA | 155 A 0.980 * |
| psbB | 494 A 0.981 * |
| psbD | 3 V 1.000 **, 4 A 1.000 **, 5 L 1.000 ** |
| rbcL | 219 L 0.999 **, 225 I 1.000 **, 226 Y 1.000 **, 240 L 1.000 **, 255 V 1.000 **, 407 L 0.998 **, 424 L 0.999 **, 449 S 1.000 ** |
| rpl20 | 69 N 0.987 *, 70 K 1.000 **, 77 R 0.984 * |
| rpl23 | 62 K 0.987 * |
| rpoB | 787 G 0.953 * |
| rpoC1 | 141 D 0.950 * |
| rpoC2 | 398 L 0.995 **, 582 S 0.957 *, 622 W 0.968 *, 673 Y 1.000 **, 707 S 1.000 **, 811 T 0.952 *, 972 I 0.987 *, 1119 W 0.990 ** |
| rps3 | 83 L 0.986 * |
| rps4 | 157 P 0.956 * |
| rps7 | 43 L 0.998 **, 51 Q 0.963 *, 81 S 1.000 **, 112 P 0.980 *, 131 S 0.994 ** |
| rps8 | 90 R 0.996 ** |
| rps15 | 72 R 0.994 ** |
| ycf3 | 109 C 0.981 *, 110 H 1.000 ** |
| ycf4 | 158 L 0.975 *, 175 R 0.976 * |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, D.-M.; Liu, H.-L.; Pan, Y.-G.; Yu, B.; Huang, D.; Zhu, G.-F. Comparative Chloroplast Genomics of 21 Species in Zingiberales with Implications for Their Phylogenetic Relationships and Molecular Dating. Int. J. Mol. Sci. 2023, 24, 15031. https://doi.org/10.3390/ijms241915031
Li D-M, Liu H-L, Pan Y-G, Yu B, Huang D, Zhu G-F. Comparative Chloroplast Genomics of 21 Species in Zingiberales with Implications for Their Phylogenetic Relationships and Molecular Dating. International Journal of Molecular Sciences. 2023; 24(19):15031. https://doi.org/10.3390/ijms241915031
Chicago/Turabian StyleLi, Dong-Mei, Hai-Lin Liu, Yan-Gu Pan, Bo Yu, Dan Huang, and Gen-Fa Zhu. 2023. "Comparative Chloroplast Genomics of 21 Species in Zingiberales with Implications for Their Phylogenetic Relationships and Molecular Dating" International Journal of Molecular Sciences 24, no. 19: 15031. https://doi.org/10.3390/ijms241915031