
Characteristics and Comparative Analysis of Seven Complete Plastomes of Trichoglottis s.l. (Aeridinae, Orchidaceae)
 , , ,
, , ,
Abstract
:1. Introduction
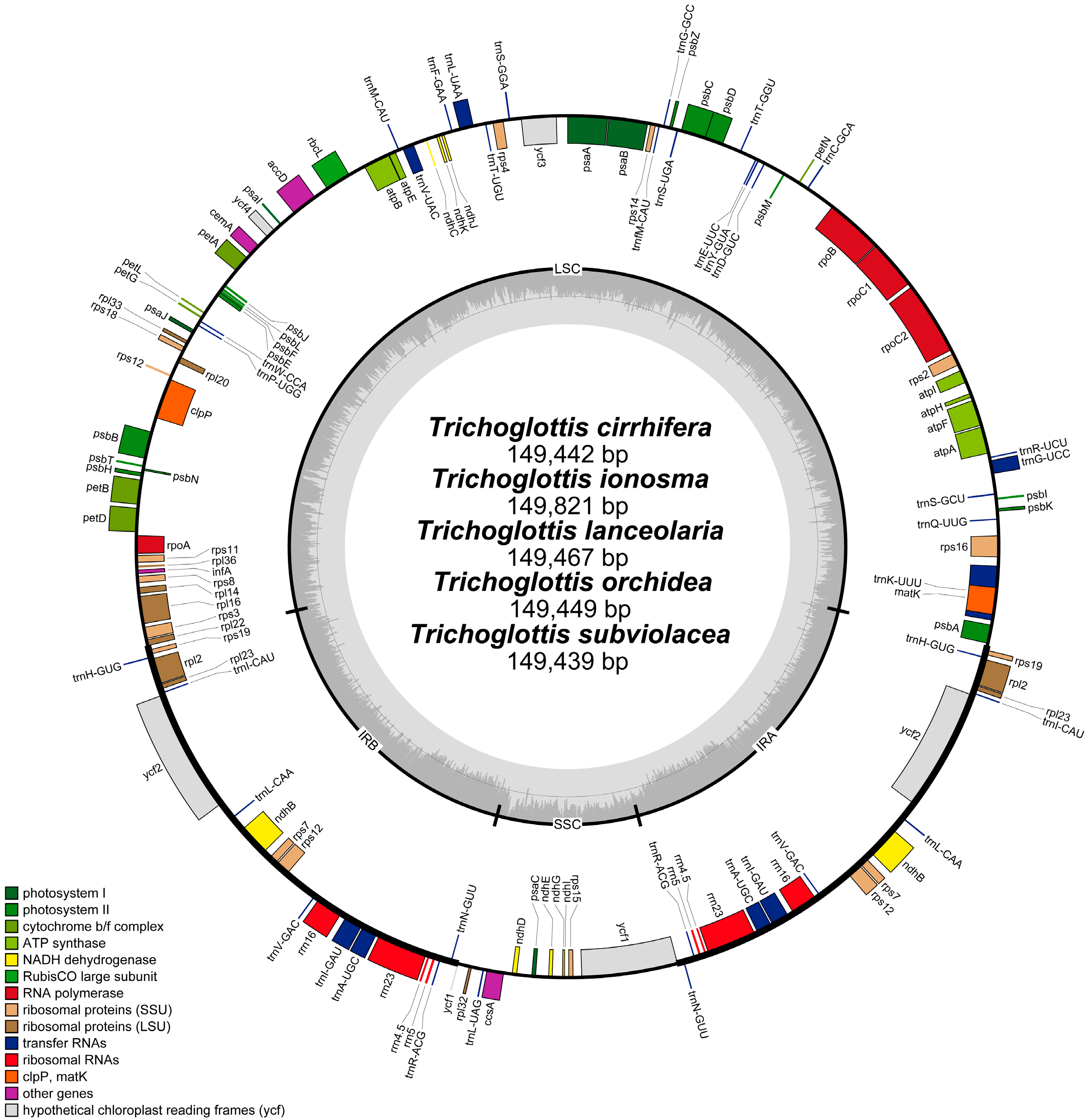
2. Results
2.1. Characteristics of the Plastome
2.2. Repeated Analysis
2.3. Codon Usage Analyses
2.4. Plastome Sequence Divergence and Barcoding Investigation
2.5. Phylogenetic Analysis
3. Discussion
3.1. The Plastome Characteristics and Structural Evolution
3.2. The Barcoding Investigation and Phylogenetic Analysis
4. Materials and Methods
4.1. Taxon Sampling and Sequencing
4.2. Plastome Assembly and Annotation
4.3. Plastome Comparative Analysis
4.4. Codon Usage Analysis
4.5. Sequence Divergence and Barcoding Investigation
4.6. Phylogenetic Reconstruction
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Blume, C.L. Bijdragen Tot de Flora van Nederlandsch Indië (Part 7); Ter Lands Drukkerij: Batavia, The Netherlands, 1825. [Google Scholar]
- Bandara, C.; Atthanagoda, A.G.; Bandara, N.L.; Ranasinghe, B.; Kumar, P. Trichoglottis longifolia (Orchidaceae: Epidendroideae: Vandeae: Aeridinae), a new species from Sri Lanka. Phytotaxa 2022, 567, 71–78. [Google Scholar] [CrossRef]
- Pridgeon, A.M.; Cribb, P.J.; Chase, M.W.; Rasmussen, F.N. Genera Orchidacearum Volume 6: Epidendroideae (Part 3); OUP Oxford Press: Oxford, UK, 2014. [Google Scholar]
- Cootes, J. Philippine Native Orchid Species; Katha Publishing: Quezon, Philippines, 2011. [Google Scholar]
- Wood, J.J.; Lamb, A.; Chan, C.L. A new species of Trichoglottis (Orchidaceae: Vandeae: Aeridinae) from Borneo. Sandaicania 1998, 11, 43–47. [Google Scholar]
- Topik, H.; Yukawa, T.; Ito, M. Molecular phylogenetics of subtribe Aeridinae (Orchidaceae): Insights from plastid matK and nuclear ribosomal ITS sequences. J. Plant Res. 2005, 118, 271–284. [Google Scholar] [CrossRef] [PubMed]
- Carlsward, B.S.; Whitten, W.M.; Williams, N.H.; Bytebier, B. Molecular phylogeny of Vandeae (Orchidaceae) and the evolution of leaflessness. Am. J. Bot. 2006, 93, 770–786. [Google Scholar] [CrossRef]
- Zou, L.H.; Huang, J.X.; Zhang, G.Q.; Liu, Z.J.; Zhuang, X.Y. A molecular phylogeny of Aeridinae (Orchidaceae: Epidendroideae) inferred from multiple nuclear and chloroplast regions. Mol. Phylogenet. Evol. 2015, 85, 247–254. [Google Scholar] [CrossRef]
- Niehuis, O.; Hartig, G.; Grath, S.; Pohl, H.; Lehmann, J.; Tafer, H.; Donath, A.; Krauss, V.; Eisenhardt, C.; Hertel, J.; et al. Genomic and morphological evidence converge to resolve the enigma of Strepsiptera. Curr. Biol. 2012, 22, 1309–1313. [Google Scholar] [CrossRef]
- Givnish, T.J.; Spalink, D.; Ames, M.; Lyon, S.P.; Hunter, S.J.; Zuluaga, A.; Iles, W.J.; Clements, M.A.; Arroyo, M.T.; Leebens-Mack, J.; et al. Orchid phylogenomics and multiple drivers of their extraordinary diversification. Proc. Biol. Sci. B 2015, 282, 2108–2111. [Google Scholar] [CrossRef]
- Ohyama, K. Chloroplast and mitochondrial genomes from a liverwort, Marchantia polymorpha—Gene organization and molecular evolution. Biosci. Biotechnol. Biochem. 1996, 60, 16–24. [Google Scholar] [CrossRef]
- Li, Y.X.; Li, Z.H.; Schuiteman, A.; Chase, M.W.; Li, J.W.; Huang, W.C.; Hidayat, A.; Wu, S.S.; Jin, X.H. Phylogenomics of Orchidaceae based on plastid and mitochondrial genomes. Mol. Phylogenet. Evol. 2019, 139, 106540. [Google Scholar] [CrossRef]
- Kim, Y.K.; Jo, S.; Cheon, S.H.; Joo, M.J.; Hong, J.R.; Kwak, M.; Kim, K.J. Plastome Evolution and Phylogeny of Orchidaceae, with 24 New Sequences. Front. Plant Sci. 2020, 11, 22. [Google Scholar] [CrossRef]
- Tu, X.D.; Liu, D.K.; Xu, S.W.; Zhou, C.Y.; Gao, X.Y.; Zeng, M.Y.; Zhang, S.; Chen, J.L.; Ma, L.; Zhou, Z.; et al. Plastid phylogenomics improves resolution of phylogenetic relationship in the Cheirostylis and Goodyera clades of Goodyerinae (Orchidoideae, Orchidaceae). Mol. Phylogenet. Evol. 2021, 164, 107269. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.K.; Tu, X.D.; Zhao, Z.; Zeng, M.Y.; Zhang, S.; Ma, L.; Zhang, G.Q.; Wang, M.M.; Liu, Z.J.; Lan, S.R.; et al. Plastid phylogenomic data yield new and robust insights into the phylogeny of Cleisostoma–Gastrochilus clades (Orchidaceae, Aeridinae). Mol. Phylogenet. Evol. 2020, 145, 106729. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.Y.; Yang, J.X.; Bai, M.Z.; Zhang, G.Q.; Liu, Z.J. The chloroplast genome evolution of Venus slipper (Paphiopedilum): IR expansion, SSC contraction, and highly rearranged SSC regions. BMC Plant Biol. 2021, 21, 248. [Google Scholar] [CrossRef]
- Schelkunov, M.I.; Shtratnikova, V.Y.; Nuraliev, M.S.; Selosse, M.A.; Penin, A.A.; Logacheva, M.D. Exploring the limits for reduction of plastid genomes: A case study of the mycoheterotrophic orchids Epipogium aphyllum and Epipogium roseum. Genome Biol. Evol. 2015, 7, 1179–1191. [Google Scholar] [CrossRef]
- Wen, Y.; Qin, Y.; Shao, B.; Li, J.; Ma, C.; Liu, Y.; Yang, B.; Jin, X. The extremely reduced, diverged and reconfigured plastomes of the largest mycoheterotrophic orchid lineage. BMC Plant Biol. 2022, 22, 448. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.L.; Wicke, S.; Li, J.W.; Han, Y.; Lin, C.S.; Li, D.Z.; Zhou, T.T.; Huang, W.C.; Huang, L.Q.; Jin, X.H. Lineage-specific reductions of plastid genomes in an orchid tribe with partially and fully mycoheterotrophic species. Genome Biol. Evol. 2016, 8, 2164–2175. [Google Scholar] [CrossRef]
- Lin, C.S.; Chen, J.J.W.; Huang, Y.T.; Chan, M.T.; Daniell, H.; Chang, W.J.; Hsu, C.T.; Liao, D.C.; Wu, F.H.; Lin, S.Y.; et al. The location and translocation of ndh genes of chloroplast origin in the Orchidaceae family. Sci. Rep. 2015, 5, 9040. [Google Scholar] [CrossRef]
- Zavala-Páez, M.; Vieira, L.D.N.; Baura, V.A.D.; Balsanelli, E.; Souza, E.M.D.; Cevallos, M.C.; Smidt, E.D.C. Comparative plastid genomics of neotropical Bulbophyllum (Orchidaceae; Epidendroideae). Front. Plant Sci. 2020, 11, 799. [Google Scholar] [CrossRef]
- Sanderson, M.J.; Copetti, D.; Burquez, A.; Bustamante, E.; Charboneau, J.L.M.; Eguiarte, L.E.; Kumar, S.; Lee, H.O.; Lee, J.; McMahon, M.; et al. Exceptional reduction of the plastid genome of saguaro cactus (Carnegiea gigantea): Loss of the ndh gene suite and inverted repeat. Am. J. Bot. 2015, 102, 1115–1127. [Google Scholar] [CrossRef]
- Niu, Z.; Zhu, S.; Pan, J.; Li, L.; Sun, J.; Ding, X. Comparative analysis of Dendrobium plastomes and utility of plastomic mutational hotspots. Sci. Rep. 2017, 7, 2073. [Google Scholar]
- Jiang, H.; Tian, J.; Yang, J.; Dong, X.; Zhong, Z.; Mwachala, G.; Zhang, C.; Hu, G.; Wang, Q. Comparative and phylogenetic analyses of six Kenya Polystachya (Orchidaceae) species based on the complete chloroplast genome sequences. BMC Plant Biol. 2022, 22, 177. [Google Scholar] [CrossRef]
- Thode, V.A.; Lohmann, L.G. Comparative Chloroplast Genomics at Low Taxonomic Levels: A Case Study Using Amphilophium (Bignonieae, Bignoniaceae). Front. Plant Sci. 2019, 10, 796. [Google Scholar] [CrossRef]
- Xi, J.; Lv, S.; Zhang, W.; Zhang, J.; Wang, K.; Guo, H.; Hu, J.; Yang, Y.; Wang, J.; Xia, G.; et al. Comparative plastomes of Carya species provide new insights into the plastomes evolution and maternal phylogeny of the genus. Front. Plant Sci. 2022, 13, 990064. [Google Scholar] [CrossRef]
- He, S.L.; Wang, Y.S.; Volis, S.; Li, D.Z.; Yi, T.S. Genetic diversity and population structure: Implications for conservation of wild soybean (Glycine soja Sieb. et Zucc) based on nuclear and chloroplast microsatellite variation. Int. J. Mol. Sci. 2012, 13, 12608–12628. [Google Scholar] [CrossRef]
- Qi, W.; Lin, F.; Liu, Y.; Huang, B.; Cheng, J.; Zhang, W.; Zhao, H. High-throughput development of simple sequence repeat markers for genetic diversity research in Crambe abyssinica. BMC Plant Biol. 2016, 16, 139. [Google Scholar] [CrossRef]
- Xiao, T.; He, L.; Yue, L.; Zhang, Y.; Lee, S.Y. Comparative phylogenetic analysis of complete plastid genomes of Renanthera (Orchidaceae). Front. Genet. 2022, 13, 998575. [Google Scholar] [CrossRef]
- Chen, Y.; Zhong, H.; Zhu, Y.; Huang, Y.; Wu, S.; Liu, Z.; Lan, S.; Zhai, J. Plastome structure and adaptive evolution of Calanthe s.l. species. PeerJ 2020, 8, e10051. [Google Scholar] [CrossRef]
- Li, L.; Wu, Q.; Fang, L.; Wu, K.; Li, M.; Zeng, S. Comparative Chloroplast Genomics and Phylogenetic Analysis of Thuniopsis and Closely Related Genera within Coelogyninae (Orchidaceae). Front. Genet. 2022, 13, 850201. [Google Scholar] [CrossRef]
- McDonald, M.J.; Wang, W.C.; Huang, H.D.; Leu, J.Y. Clusters of nucleotide substitutions and insertion/deletion mutations are associated with repeat sequences. PLoS Biol. 2011, 9, e1000622. [Google Scholar] [CrossRef]
- Gragg, H.; Harfe, B.D.; Jinks-Robertson, S. Base composition of mononucleotide runs affects DNA polymerase slippage and removal of frameshift intermediates by mismatch repair in Saccharomyces cerevisiae. Mol. Cell. Biol. 2002, 22, 8756–8762. [Google Scholar] [CrossRef]
- Yang, X.; Luo, X.; Cai, X. Analysis of codon usage pattern in Taenia saginata based on a transcriptome dataset. Parasites Vectors 2014, 7, 527. [Google Scholar] [CrossRef]
- Knill, T.; Reichelt, M.; Paetz, C.; Gershenzon, J.; Binder, S. Arabidopsis Thaliana Encodes a Bacterial-Type Heterodimeric Isopropylmalate Isomerase Involved in Both Leu Biosynthesis and the Met Chain Elongation Pathway of Glucosinolate Formation. Plant Mol. Biol. 2009, 71, 227–239. [Google Scholar] [CrossRef]
- Hildebrandt, T.M.; Nesi, A.N.; Araújo, W.L.; Braun, H.P. Amino Acid Catabolism in Plants. Mol. Plant 2015, 8, 1563–1579. [Google Scholar] [CrossRef]
- Leaché, A.D.; Banbury, B.L.; Linkem, C.W.; De Oca, A.N.-M. Phylogenomics of a rapid radiation: Is chromosomal evolution linked to increased diversification in north american spiny lizards (Genus Sceloporus)? BMC Evol. Biol. 2016, 16, 63. [Google Scholar] [CrossRef]
- Jin, J.-J.; Yu, W.-B.; Yang, J.-B.; Song, Y.; DePamphilis, C.W.; Yi, T.S.; Li, D.Z. GetOrganelle: A fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome Biol. 2020, 21, 241. [Google Scholar] [CrossRef]
- Qu, X.J.; Moore, M.J.; Li, D.Z.; Yi, T.S. PGA: A software package for rapid, accurate, and flexible batch annotation of plastomes. Plant Methods 2019, 15, 50. [Google Scholar] [CrossRef]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef]
- Greiner, S.; Lehwark, P.; Bock, R. OrganellarGenomeDRAW (OGDRAW) version 1.3.1: Expanded toolkit for the graphical visualization of organellar genomes. Nucleic Acids Res. 2019, 47, W59–W64. [Google Scholar] [CrossRef]
- Beier, S.; Thiel, T.; Münch, T.; Scholz, U.; Mascher, M. MISA-web: A web server for microsatellite prediction. Bioinformatics 2017, 33, 2583–2585. [Google Scholar] [CrossRef] [PubMed]
- Kurtz, S.; Choudhuri, J.V.; Ohlebusch, E.; Schleiermacher, C.; Stoye, J.; Giegerich, R. REPuter: The manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 2001, 29, 4633–4642. [Google Scholar] [CrossRef] [PubMed]
- Villanueva, R.A.M.; Chen, Z.J. ggplot2: Elegant Graphics for Data Analysis (2nd Ed.). Meas. Interdiscip. Res. Perspect. 2019, 17, 160–167. [Google Scholar] [CrossRef]
- Rissman, A.I.; Mau, B.; Biehl, B.S.; Darling, A.E.; Glasner, J.D.; Perna, N.T. Reordering contigs of draft genomes using the Mauve aligner. Bioinformatics 2009, 25, 2071–2073. [Google Scholar] [CrossRef]
- Amiryousefi, A.; Hyvonen, J.; Poczai, P. IRscope: An online program to visualize the junction sites of chloroplast genomes. Bioinformatics 2018, 34, 3030–3031. [Google Scholar] [CrossRef]
- Zhang, D.; Gao, F.; Jakovlić, I.; Zou, H.; Zhang, J.; Li, W.X.; Wang, G.T. PhyloSuite: An integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol. Ecol. Resour. 2020, 20, 348–355. [Google Scholar] [CrossRef]
- Xia, X. Dambe7: New and improved tools for data analysis in molecular biology and evolution. Mol. Biol. Evol. 2018, 35, 1550–1552. [Google Scholar] [CrossRef]
- Chen, C.J.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.H.; Xia, R. TBtools: An integrative toolkit developed for interactive analyses of big biological data. Mol. Plant 2020, 13, 1194–1202. [Google Scholar] [CrossRef]
- Brudno, M.; Malde, S.; Poliakov, A.; Do, C.B.; Couronne, O.; Dubchak, I.; Batzoglou, S. Glocal alignment: Finding rearrangements during alignment. Bioinformatics 2003, 19, i54–i62. [Google Scholar] [CrossRef]
- Rozas, J.; Sánchez-DelBarrio, J.C.; Messeguer, X.; Rozas, R. Dnasp, dna polymorphism analyses by the coalescent and other methods. Bioinformatics 2003, 19, 2496–2497. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Capella-Gutierrez, S.; Silla-Martinez, J.M.; Gabaldon, T. Trimal: A tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 2009, 25, 1972–1973. [Google Scholar] [CrossRef]
- Miller, M.A.; Pfeiffer, W.; Schwartz, T. Creating the CIPRES Science Gateway for inference of large phylogenetic trees. In Proceedings of the 2010 Gateway Computing Environments Workshop (GCE), New Orleans, LA, USA, 14 November 2010; pp. 1–8. [Google Scholar]
- Stamatakis, A.; Hoover, P.; Rougemont, J. A rapid bootstrap algorithm for the RAxML Web servers. Syst. Biol. 2008, 57, 758–771. [Google Scholar] [CrossRef] [PubMed]
- Felsenstein, J. Confidence limits on phylogenies: An approach using the bootstrap. Evol. Int. J. Org. Evol. 1985, 39, 783–791. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species Name | Size (bp) | GC Content (%) | LSC Size in bp (%) | IR Size in bp (%) | SSC Size in bp (%) | Total Number of Gene | Protein-Encoding Gene | tRNA | rRNA | Number of ndh Fragment |
|---|---|---|---|---|---|---|---|---|---|---|
| T. cirrhifera * | 149,442 | 36.7 | 85,721 (57.36) | 25,775 (17.25) | 12,171 (8.14) | 120 | 74 | 38 | 8 | 8 |
| T. ionosma * | 149,841 | 36.6 | 86,210 (57.53) | 25,812 (17.23) | 12,007 (8.01) | 120 | 74 | 38 | 8 | 7 |
| T. lanceolaria * | 149,467 | 36.7 | 85,728 (57.36) | 25,784 (17.25) | 12,171 (8.14) | 120 | 74 | 38 | 8 | 8 |
| T. latisepala | 149,402 | 36.7 | 85,681 (57.35) | 25,784 (17.26) | 12,153 (8.13) | 120 | 74 | 38 | 8 | 8 |
| T. orchidea * | 149,449 | 36.7 | 85,715 (57.35) | 25,782 (17.25) | 12,170 (8.14) | 120 | 74 | 38 | 8 | 8 |
| T. philippinensis | 149,663 | 36.7 | 86,069 (57.51) | 25,784 (17.23) | 12,026 (8.04) | 120 | 74 | 38 | 8 | 8 |
| T. subviolacea * | 149,439 | 36.7 | 85,853 (57.45) | 25,791 (17.26) | 12,004 (8.03) | 120 | 74 | 38 | 8 | 8 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, C.-Y.; Zeng, M.-Y.; Gao, X.; Zhao, Z.; Li, R.; Wu, Y.; Liu, Z.-J.; Zhang, D.; Li, M.-H. Characteristics and Comparative Analysis of Seven Complete Plastomes of Trichoglottis s.l. (Aeridinae, Orchidaceae). Int. J. Mol. Sci. 2023, 24, 14544. https://doi.org/10.3390/ijms241914544
Zhou C-Y, Zeng M-Y, Gao X, Zhao Z, Li R, Wu Y, Liu Z-J, Zhang D, Li M-H. Characteristics and Comparative Analysis of Seven Complete Plastomes of Trichoglottis s.l. (Aeridinae, Orchidaceae). International Journal of Molecular Sciences. 2023; 24(19):14544. https://doi.org/10.3390/ijms241914544
Chicago/Turabian StyleZhou, Cheng-Yuan, Meng-Yao Zeng, Xuyong Gao, Zhuang Zhao, Ruyi Li, Yuhan Wu, Zhong-Jian Liu, Diyang Zhang, and Ming-He Li. 2023. "Characteristics and Comparative Analysis of Seven Complete Plastomes of Trichoglottis s.l. (Aeridinae, Orchidaceae)" International Journal of Molecular Sciences 24, no. 19: 14544. https://doi.org/10.3390/ijms241914544