

From Plant to Chemistry: Sources of Active Opioid Antinociceptive Principles for Medicinal Chemistry and Drug Design
 , , , , and
, , , , and
Abstract
: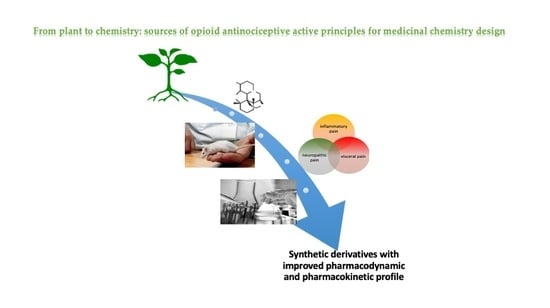
1. Introduction
2. Method Section
3. Sources of Active Opioid Antinociceptive Principles
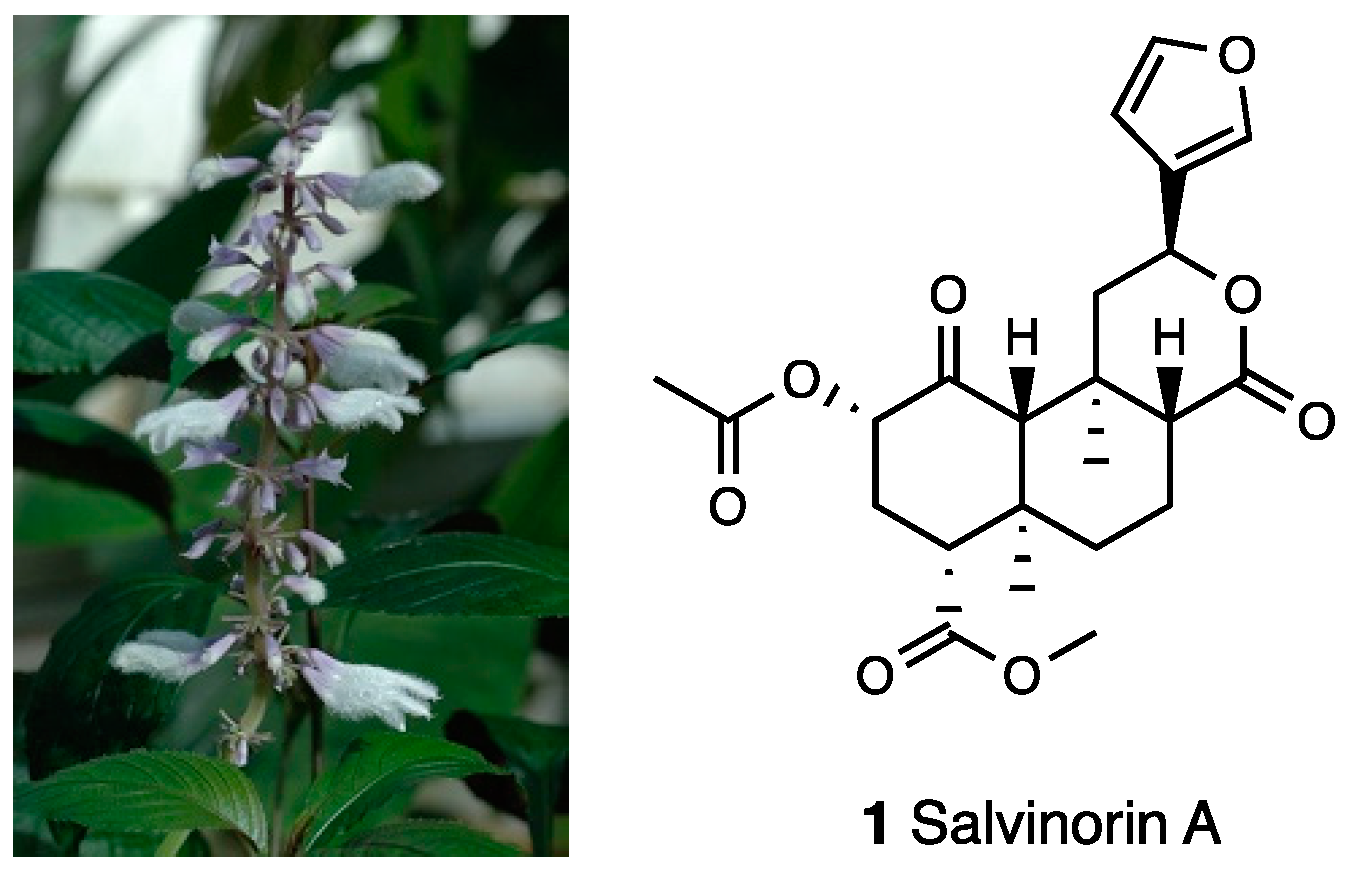
3.1. Salvinorin A
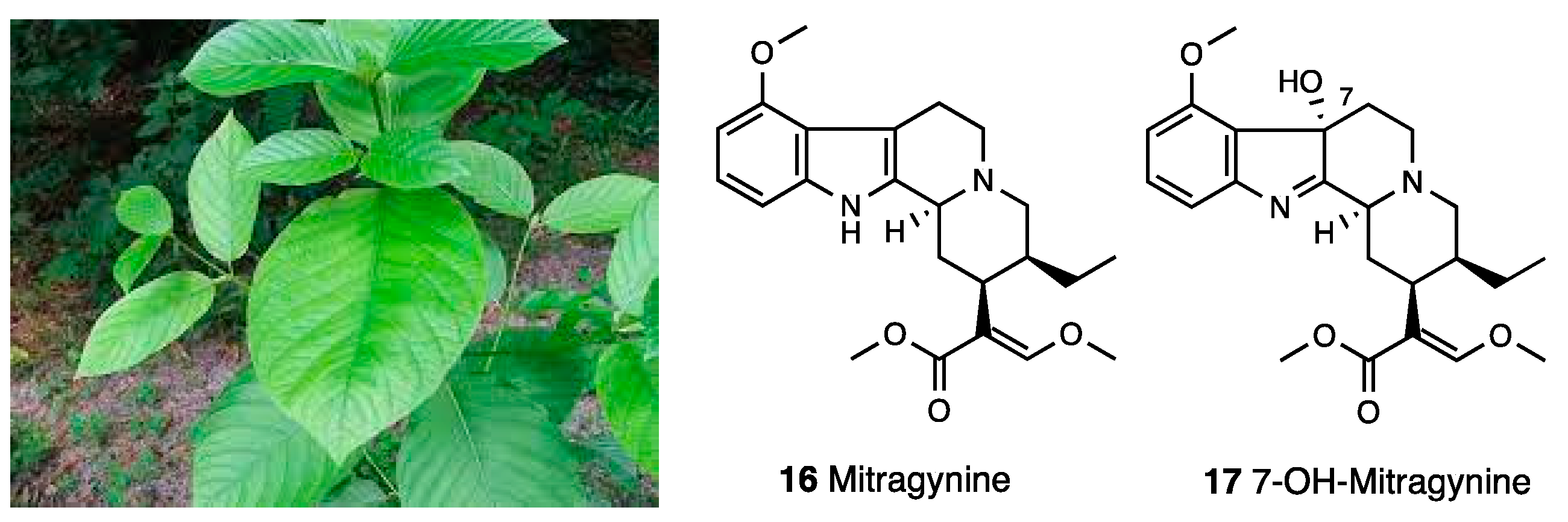
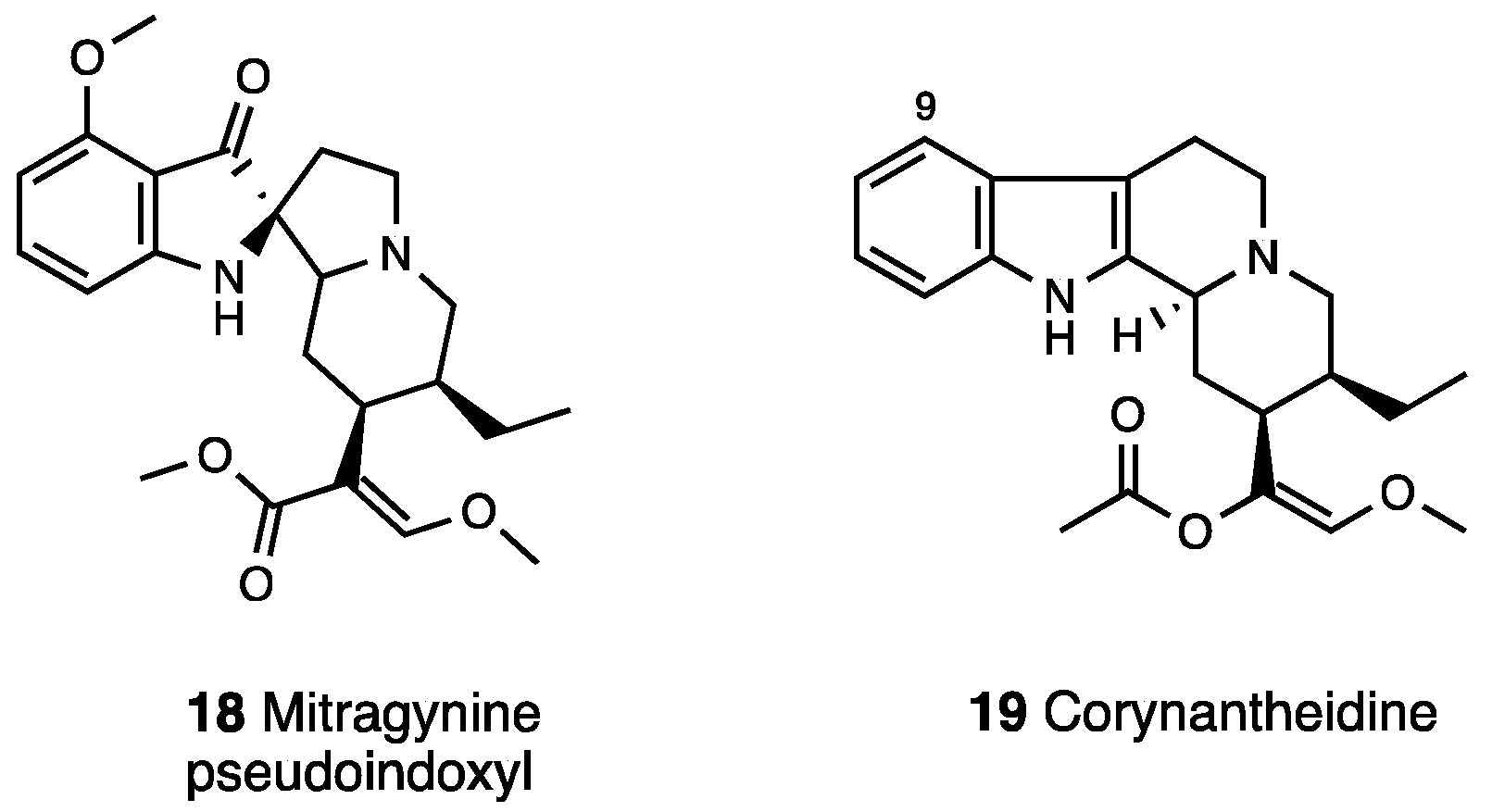
3.2. Mitragynine
3.3. Collybolide
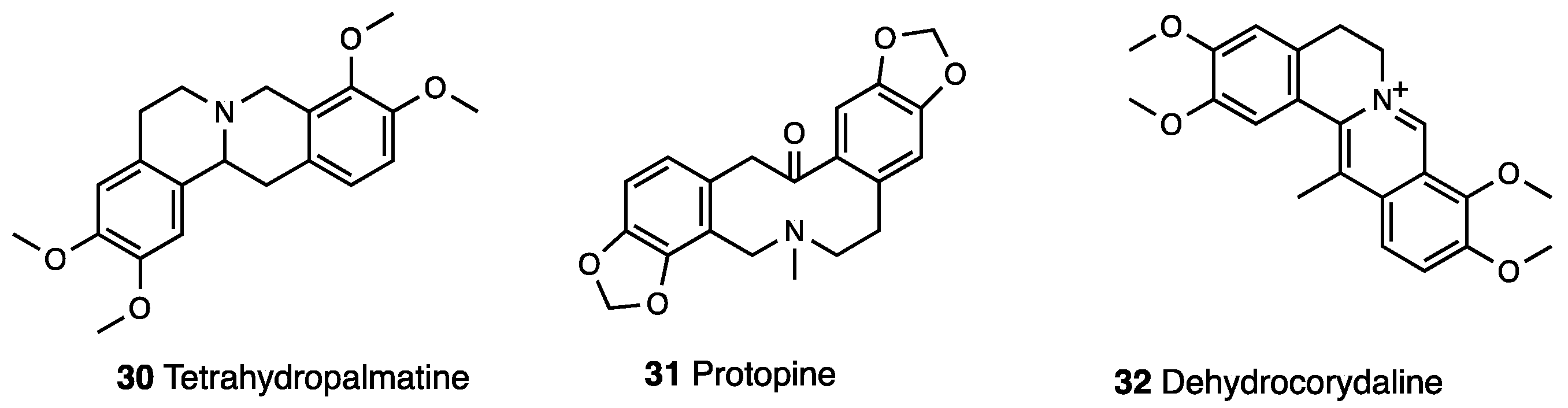
3.4. Corydine, Corydaline, Dehydrocorybulbine (DHCB)
3.5. Essential Oil of Himenaea cangaceira
3.6. Polymethoxyflavones of Ageratum conyzoides
3.7. Brachydin A, Brachydin B, and Brachydin C
3.8. Nuciferine and N-Nornuciferine
3.9. Clinacanthus nutans
3.10. Azadirachta indica Extracts and Oleum Azadirachti
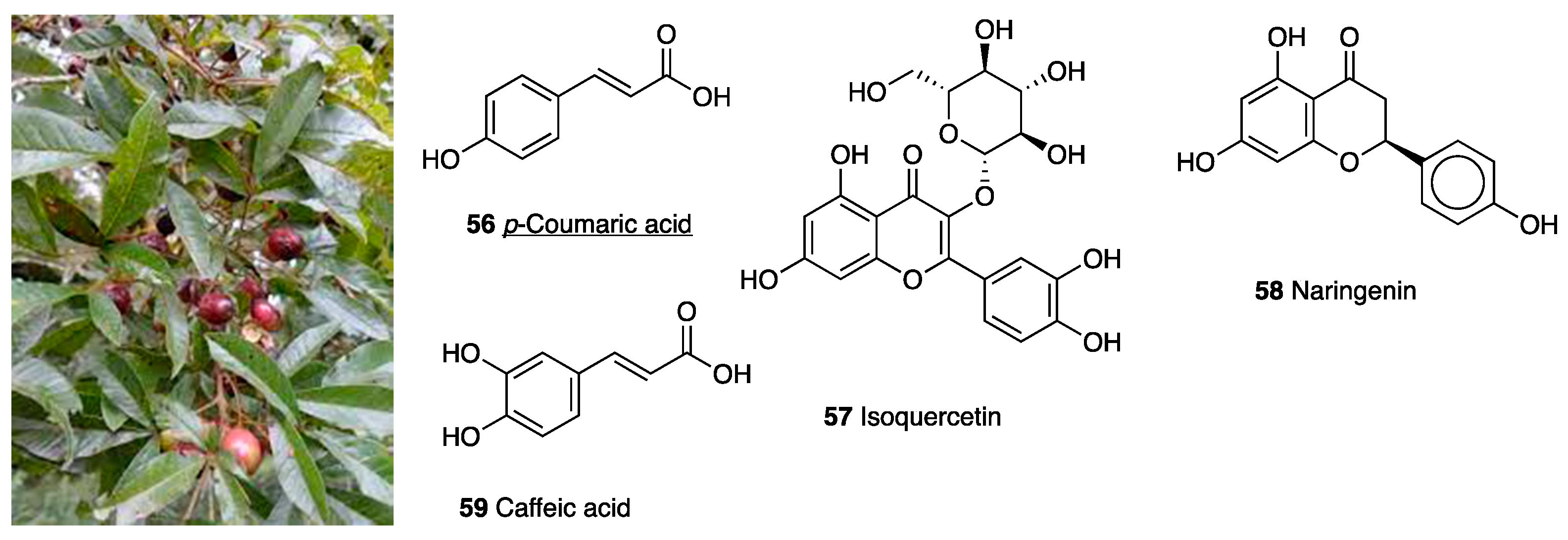
3.11. Vitex megapotamica Extracts
3.12. (–)-Hardwickiic Acid
3.13. Algrizea minor Essential Oil
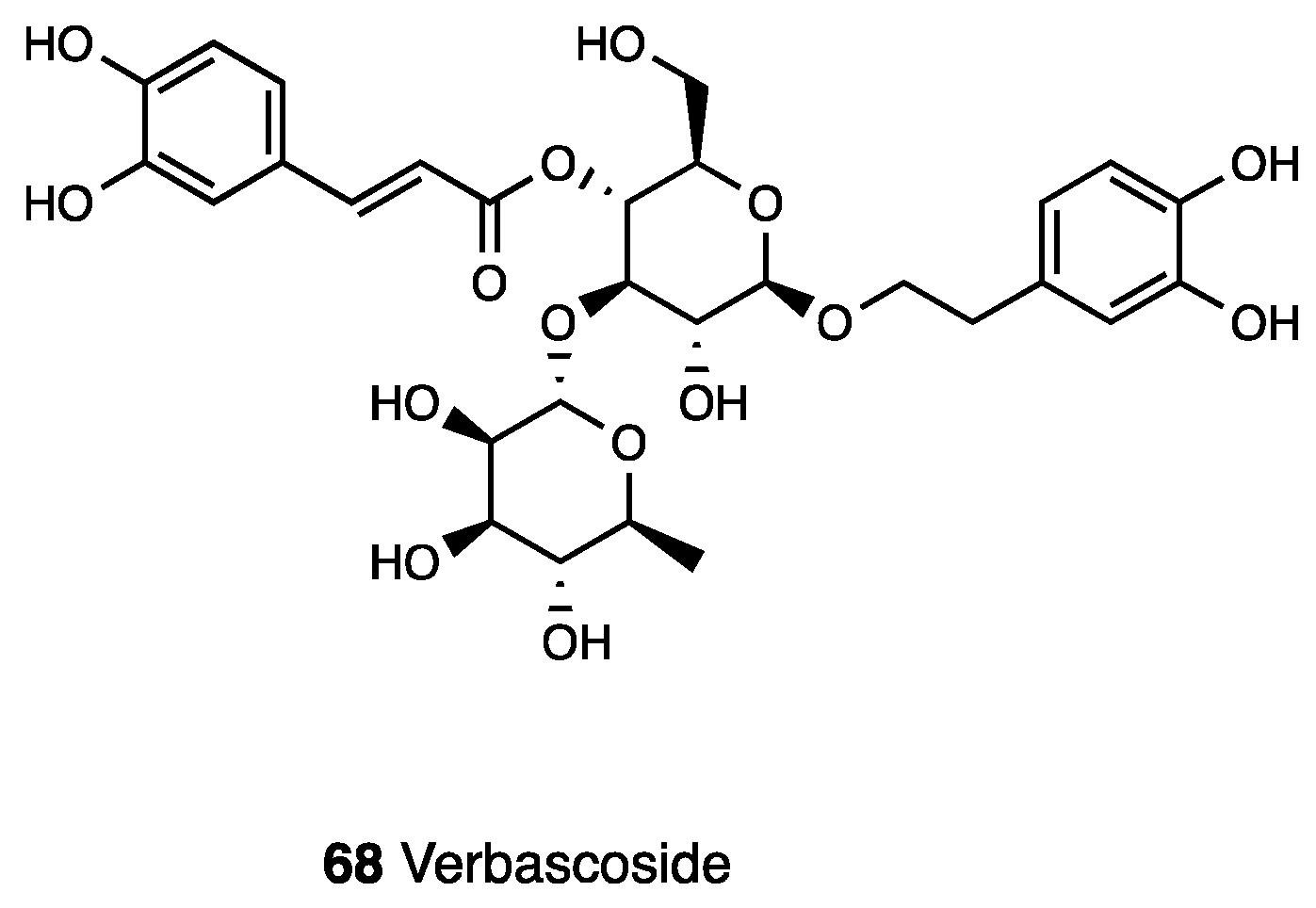
3.14. Verbascoside
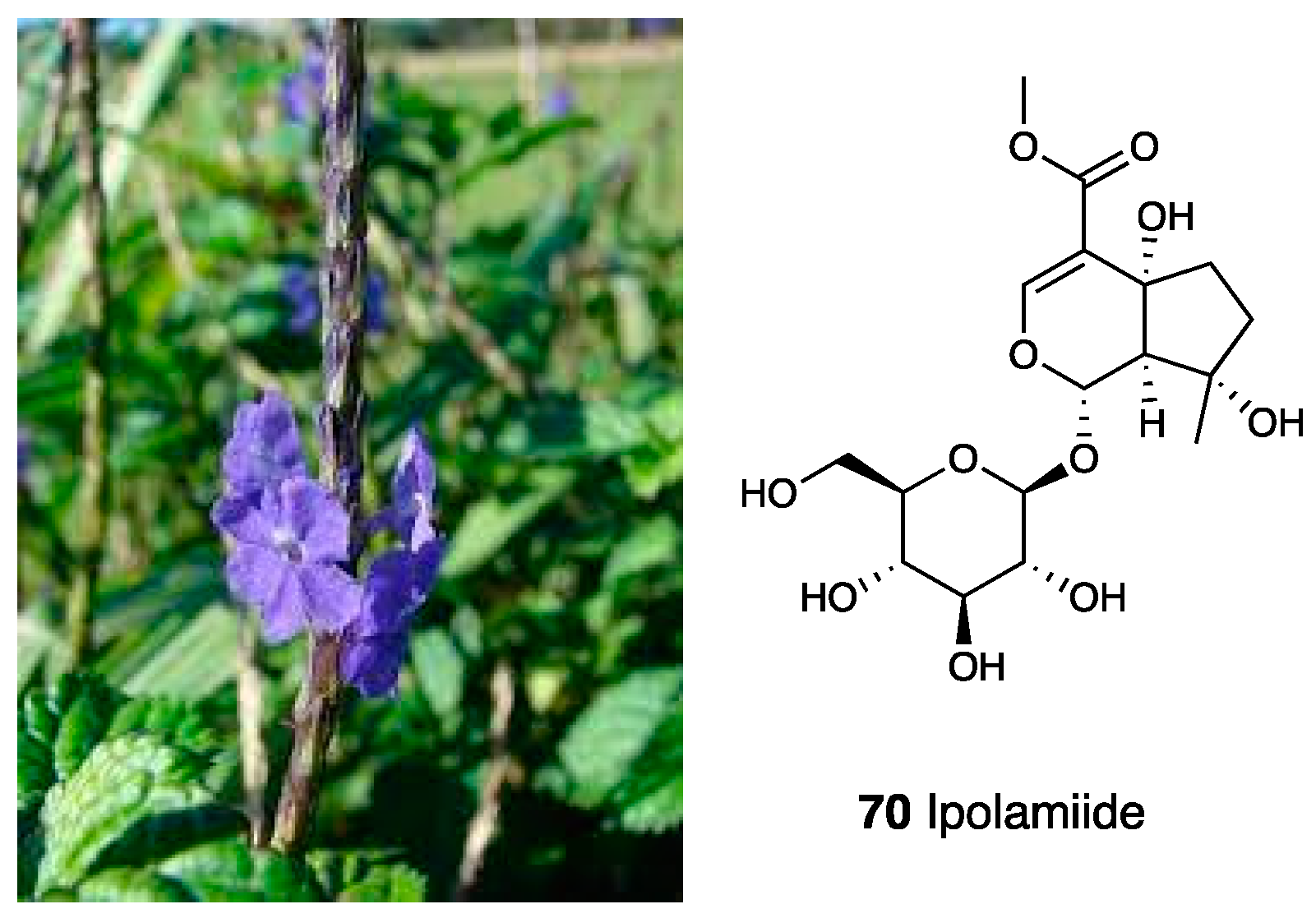
3.15. Aqueous Extracts of Stachytarpheta cayennensis
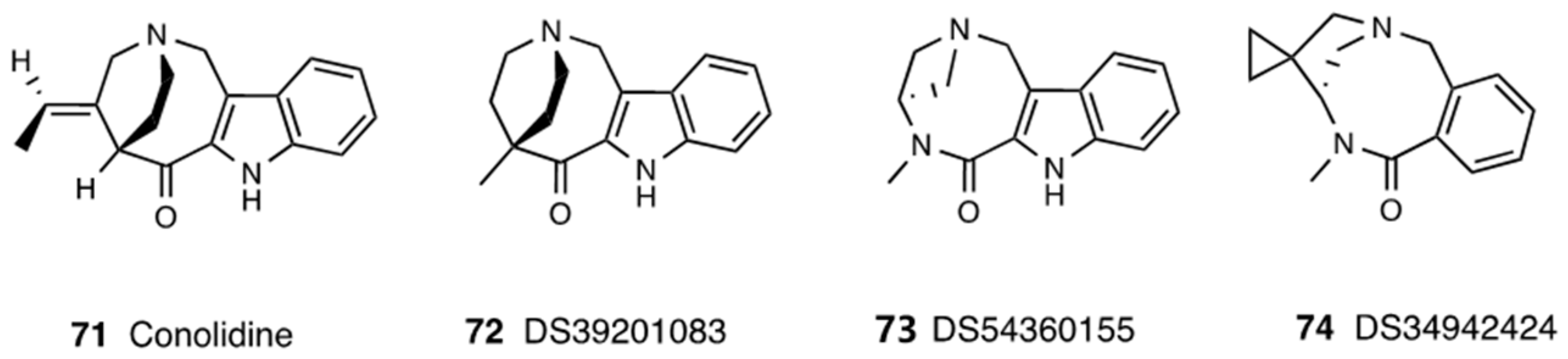
3.16. Conolidine
3.17. Rubiscolin
3.18. Tingenone
3.19. Esters of N-Methylanthranilic Acid
3.20. Mansoa alliacea Extract
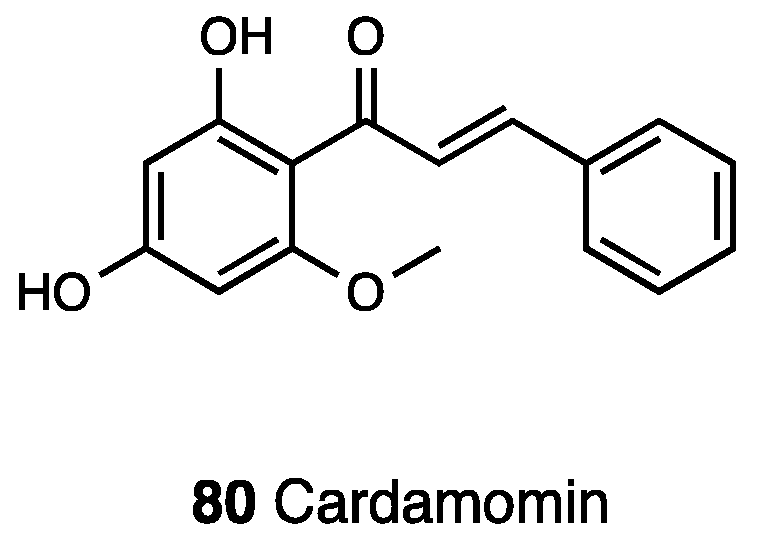
3.21. Cardamonin
3.22. Berberine
3.23. Lappaconitine
3.24. Oleanolic Acid
3.25. Urundeuvine A, B and C
3.26. Artemisia annua L.
3.27. Sitosterole and Wrightiadione of Wrightia coccinea
3.28. Thymoquinone
3.29. (–)-Linalool
3.30. Zerumbone
3.31. Salvia officinalis
4. Discussion and Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Fidan, O.; Ren, J.; Zhan, J. Engineered production of bioactive natural products from medicinal plants. World J. Tradit. Chin. Med. 2022, 8, 59–76. [Google Scholar] [CrossRef]
- Yñigez-Gutierrez, A.E.; Bachmann, B.O. Fixing the Unfixable: The Art of Optimizing Natural Products for Human Medicine. J. Med. Chem. 2019, 62, 8412–8428. [Google Scholar] [CrossRef] [PubMed]
- Pitchai, D.; Manikkam, R.; Rajendran, S.R.; Pitchai, G. Database on pharmacophore analysis of active principles, from medicinal plants. Bioinformation 2010, 5, 43–45. [Google Scholar] [CrossRef] [PubMed]
- Chopra, B.; Dhingra, A.K. Natural products: A lead for drug discovery and development. Phytother. Res. 2021, 35, 4660–4702. [Google Scholar] [CrossRef]
- Gallego-Jara, J.; Lozano-Terol, G.; Sola-Martínez, R.A.; Cánovas-Díaz, M.; de Diego Puente, T. A Compressive Review about Taxol®: History and Future Challenges. Molecules 2020, 25, 5986. [Google Scholar] [CrossRef]
- Brennan, M.S.; Matos, M.F.; Li, B.; Hronowski, X.; Gao, B.; Juhasz, P.; Rhodes, K.J.; Scannevin, R.H. Dimethyl fumarate and monoethyl fumarate exhibit differential effects on KEAP1, NRF2 activation, and glutathione depletion in vitro. PLoS ONE 2015, 10, e0120254. [Google Scholar] [CrossRef]
- Brück, J.; Dringen, R.; Amasuno, A.; Pau-Charles, I.; Ghoreschi, K. A review of the mechanisms of action of dimethylfumarate in the treatment of psoriasis. Exp. Dermatol. 2018, 27, 611–624. [Google Scholar] [CrossRef]
- Walker, F.; Adamczyk, A.; Kellerer, C.; Belge, K.; Brück, J.; Berner, T.; Merten, K.; Núnez Gómez, N.; Neureither, M.; Röcken, M.; et al. Study group. Fumaderm® in daily practice for psoriasis: Dosing, efficacy and quality of life. Br. J. Dermatol. 2014, 171, 1197–1205. [Google Scholar] [CrossRef]
- Blair, H.A. Dimethyl Fumarate: A Review in Relapsing-Remitting MS. Drugs 2019, 79, 1965–1976. [Google Scholar] [CrossRef]
- Boye, A.; Addo, J.K.; Acheampong, D.O.; Thomford, A.K.; Asante, E.; Amoaning, R.E.; Kuma, D.N. The hydroxyl moiety on carbon one (C1) in the monoterpene nucleus of thymol is indispensable for anti-bacterial effect of thymol. Heliyon 2020, 6, e03492. [Google Scholar] [CrossRef]
- Wicks, C.; Hudlicky, T.; Rinner, U. Morphine alkaloids: History, biology, and synthesis. Alkaloids Chem. Biol. 2021, 86, 145–342. [Google Scholar] [PubMed]
- Paul, B.; Sribhashyam, S.; Majumdar, S. Opioid signaling and design of analgesics. Prog. Mol. Biol. Transl. Sci. 2023, 195, 153–176. [Google Scholar] [PubMed]
- Turnaturi, R.; Aricò, G.; Ronsisvalle, G.; Parenti, C.; Pasquinucci, L. Multitarget opioid ligands in pain relief: New players in an old game. Eur. J. Med. Chem. 2016, 108, 211–228. [Google Scholar] [CrossRef] [PubMed]
- Turnaturi, R.; Arico, G.; Ronsisvalle, G.; Pasquinucci, L.; Parenti, C. Multitarget Opioid/Non-Opioid Ligands: A Potential Approach in Pain Management. Curr. Med. Chem. 2016, 23, 4506–4528. [Google Scholar] [CrossRef]
- Al-Hasani, R.; Bruchas, M.R. Molecular mechanisms of opioid receptor-dependent signaling and behavior. Anesthesiology 2011, 115, 1363–1381. [Google Scholar] [CrossRef]
- Turnaturi, R.; Chiechio, S.; Salerno, L.; Rescifina, A.; Pittalà, V.; Cantarella, G.; Tomarchio, E.; Parenti, C.; Pasquinucci, L. Progress in the development of more effective and safer analgesics for pain management. Eur. J. Med. Chem. 2019, 183, 111701. [Google Scholar] [CrossRef]
- Atanasov, A.G.; Zotchev, S.B.; Dirsch, V.M.; Supuran, C.T. Natural products in drug discovery: Advances and opportunities. Nat. Rev. Drug Discov. 2021, 20, 200–216. [Google Scholar] [CrossRef]
- Hanes, K.R. Salvia divinorum: Clinical and Research Potential. Maps 2003, 13, 18–20. [Google Scholar]
- Ortega, A.; Blount, J.F.; Manchand, P.S. Salvinorin, a new trans-neoclerodane diterpene from Salvia divinorum (Labiatae). J. Chem. Soc. Perkin Trans. 1 1982, 1, 2505–2508. [Google Scholar] [CrossRef]
- Roth, B.L.; Baner, K.; Westkaemper, R.; Siebert, D.; Rice, K.C.; Steinberg, S.; Ernsberger, P.; Rothman, R.B. Salvinorin A: A potent naturally occurring nonnitrogenous κ opioid selective agonist. Proc. Natl. Acad. Sci. USA 2002, 99, 11934–11939. [Google Scholar] [CrossRef]
- Yan, F.; Roth, B.L. Salvinorin A: A novel and highly selective κ-opioid receptor agonist. Life Sci. 2004, 75, 2615–2619. [Google Scholar] [CrossRef] [PubMed]
- Rothman, R.B.; Murphy, D.L.; Xu, H.; Godin, J.A.; Dersch, C.M.; Partilla, J.S.; Tidgewell, K.; Schmidt, M.; Prisinzano, T.E. Salvinorin A: Allosteric interactions at the mu-opioid receptor. J. Pharmacol. Exp. Ther. 2007, 320, 801–810. [Google Scholar] [CrossRef] [PubMed]
- John, T.F.; French, L.G.; Erlichman, J.S. The antinociceptive effect of Salvinorin A in mice. Eur. J. Pharmacol. 2006, 545, 129–133. [Google Scholar] [CrossRef]
- McCurdy, C.R.; Sufka, K.J.; Smith, G.H.; Warnick, J.E.; Nieto, M.J. Antinociceptive profile of Salvinorin A, a structurally unique kappa opioid receptor agonist. Pharmacol. Biochem. Behav. 2006, 83, 109–113. [Google Scholar] [CrossRef]
- Wang, Y.; Tang, K.; Inan, S.; Siebert, D.; Holzgrabe, U.; Lee, D.Y.; Huang, P.; Li, J.G.; Cowan, A.; Liu-Chen, L.Y. Comparison of pharmacological activities of three distinct kappa ligands (Salvinorin A, TRK-820 and 3FLB) on kappa opioid receptors in vitro and their antipruritic and antinociceptive activities in vivo. J. Pharmacol. Exp. Ther. 2005, 312, 220–230. [Google Scholar] [CrossRef] [PubMed]
- Chavkin, C.; Sud, S.; Jin, W.; Stewart, J.; Zjawiony, J.K.; Siebert, D.J.; Toth, B.A.; Hufeisen, S.J.; Roth, B.L. Salvinorin A, an active component of the hallucinogenic sage Salvia divinorum is a highly efficacious κ-opioid receptor agonist: Structural and functional considerations. J. Pharmacol. Exp. Ther. 2004, 308, 1197–1203. [Google Scholar] [CrossRef] [PubMed]
- Munro, T.A.; Rizzacasa, M.A.; Roth, B.L.; Toth, B.A.; Yan, F. Studies toward the pharmacophore of Salvinorin A, a potent κ opioid receptor agonist. J. Med. Chem. 2005, 48, 345–348. [Google Scholar] [CrossRef]
- Ma, Z.; Lee, D.Y. Synthesis of deacetyl-1,10-didehydrosalvinorin G. Tetrahedron Lett. 2008, 49, 1782–1785. [Google Scholar] [CrossRef]
- Lee, D.Y.; Ma, Z.; Liu-Chen, L.Y.; Wang, Y.; Chen, Y.; Carlezon, W.A., Jr.; Cohen, B. New neoclerodane diterpenoids isolated from the leaves of Salvia divinorum and their binding affinities for human kappa opioid receptors. Bioorg. Med. Chem. 2005, 13, 5635–5639. [Google Scholar] [CrossRef]
- Béguin, C.; Richards, M.R.; Wang, Y.; Chen, Y.; Liu-Chen, L.Y.; Ma, Z.; Lee, D.Y.; Carlezon, W.A., Jr.; Cohen, B.M. Synthesis and in vitro pharmacological evaluation of Salvinorin A analogues modified at C(2). Bioorg. Med. Chem. Lett. 2005, 15, 2761–2765. [Google Scholar] [CrossRef]
- Simpson, D.S.; Katavic, P.L.; Lozama, A.; Harding, W.W.; Parrish, D.; Deschamps, J.R.; Dersch, C.M.; Partilla, J.S.; Rothman, R.B.; Navarro, H.; et al. Synthetic studies of neoclerodane diterpenes from Salvia divinorum: Preparation and opioid receptor activity of salvinicin analogues. J. Med. Chem. 2007, 50, 3596–3603. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, C.W.; Rothman, R.B.; Prisinzano, T.E. Neuropharmacology of the naturally occurring κ-opioid hallucinogen salvinorin A. Pharmacol. Rev. 2011, 63, 316–347. [Google Scholar] [CrossRef] [PubMed]
- Prisinzano, T.E.; Rothman, R.B. Salvinorin A analogs as probes in opioid pharmacology. Chem. Rev. 2008, 108, 1732–1743. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.Y.; Karnati, V.V.; He, M.; Liu-Chen, L.Y.; Kondaveti, L.; Ma, Z.; Wang, Y.; Chen, Y.; Beguin, C.; Carlezon, W.A., Jr.; et al. Synthesis and in vitro pharmacological studies of new C(2) modified salvinorin A analogues. Bioorg. Med. Chem. Lett. 2005, 15, 3744–3747. [Google Scholar] [CrossRef] [PubMed]
- Morani, A.S.; Ewald, A.; Prevatt-Smith, K.M.; Prisinzano, T.E.; Kivell, B.M. The 2-methoxy methyl analogue of salvinorin A attenuates cocaine-induced drug seeking and sucrose reinforcements in rats. Eur. J. Pharmacol. 2013, 720, 69–76. [Google Scholar] [CrossRef]
- Simonson, B.; Morani, A.S.; Ewald, A.W.; Walker, L.; Kumar, N.; Simpson, D.; Miller, J.H.; Prisinzano, T.E.; Kivell, B.M. Pharmacology and anti-addiction effects of the novel κ opioid receptor agonist Mesyl Sal B, a potent and long-acting analogue of salvinorin A. Br. J. Pharmacol. 2015, 172, 515–531. [Google Scholar] [CrossRef]
- Yan, F.; Bikbulatov, R.V.; Mocanu, V.; Dicheva, N.; Parker, C.E.; Wetsel, W.C.; Mosier, P.D.; Westkaemper, R.B.; Allen, J.A.; Zjawiony, J.K.; et al. Structure-based design, synthesis, and biochemical and pharmacological characterization of novel salvinorin A analogues as active state probes of the κ-opioid receptor. Biochemistry 2009, 48, 6898–6908. [Google Scholar] [CrossRef]
- White, K.L.; Scopton, A.P.; Rives, M.L.; Bikbulatov, R.V.; Polepally, P.R.; Brown, P.J.; Kenakin, T.; Javitch, J.A.; Zjawiony, J.K.; Roth, B.L. Identification of novel functionally selective κ-opioid receptor scaffolds. Mol. Pharmacol. 2014, 85, 83–90. [Google Scholar] [CrossRef]
- Polepally, P.R.; Huben, K.; Vardy, E.; Setola, V.; Mosier, P.D.; Roth, B.L.; Zjawiony, J.K. Michael acceptor approach to the design of new salvinorin A-based high affinity ligands for the κ-opioid receptor. Eur. J. Med. Chem. 2014, 85, 818–829. [Google Scholar] [CrossRef]
- Sałaga, M.; Polepally, P.R.; Sobczak, M.; Grzywacz, D.; Kamysz, W.; Sibaev, A.; Storr, M.; Do Rego, J.C.; Zjawiony, J.K.; Fichna, J. Novel orally available salvinorin A analog PR-38 inhibits gastrointestinal motility and reduces abdominal pain in mouse models mimicking irritable bowel syndrome. J. Pharmacol. Exp. Ther. 2014, 350, 69–78. [Google Scholar] [CrossRef]
- Sałaga, M.; Polepally, P.R.; Zakrzewski, P.K.; Cygankiewicz, A.; Sobczak, M.; Kordek, R.; Zjawiony, J.K.; Krajewska, W.M.; Fichna, J. Novel orally available salvinorin A analog PR-38 protects against experimental colitis and reduces abdominal pain in mice by interaction with opioid and cannabinoid receptors. Biochem. Pharmacol. 2014, 92, 618–626. [Google Scholar] [CrossRef] [PubMed]
- Salaga, M.; Polepally, P.R.; Zielinska, M.; Marynowski, M.; Fabisiak, A.; Murawska, N.; Sobczak, K.; Sacharczuk, M.; Do Rego, J.C.; Roth, B.L.; et al. Salvinorin A analogues PR-37 and PR-38 attenuate compound 48/80-induced itch responses in mice. Br. J. Pharmacol. 2015, 172, 4331–4341. [Google Scholar] [CrossRef] [PubMed]
- Prevatt-Smith, K.M.; Lovell, K.M.; Simpson, D.S.; Day, V.W.; Douglas, J.T.; Bosch, P.; Dersch, C.M.; Rothman, R.B.; Kivell, B.; Prisinzano, T.E. Potential Drug Abuse Therapeutics Derived from the Hallucinogenic Natural Product Salvinorin A. MedChemComm 2011, 2, 1217–1222. [Google Scholar] [CrossRef] [PubMed]
- Harding, W.W.; Tidgewell, K.; Byrd, N.; Cobb, H.; Dersch, C.M.; Butelman, E.R.; Rothman, R.B.; Prisinzano, T.E. Neoclerodane diterpenes as a novel scaffold for mu opioid receptor ligands. J. Med. Chem. 2005, 48, 4765–4771. [Google Scholar] [CrossRef]
- Groer, C.E.; Tidgewell, K.; Moyer, R.A.; Harding, W.W.; Rothman, R.B.; Prisinzano, T.E.; Bohn, L.M. An opioid agonist that does not induce μ-opioid receptor-arrestin interactions or receptor internalization. Mol. Pharmacol. 2007, 71, 549–557. [Google Scholar] [CrossRef]
- Tidgewell, K.; Harding, W.W.; Lozama, A.; Cobb, H.; Shah, K.; Kannan, P.; Dersch, C.M.; Parrish, D.; Deschamps, J.R.; Rothman, R.B.; et al. Synthesis of salvinorin A analogues as opioid receptor probes. J. Nat. Prod. 2006, 69, 914–918. [Google Scholar] [CrossRef]
- Tidgewell, K.; Groer, C.E.; Harding, W.W.; Lozama, A.; Schmidt, M.; Marquam, A.; Hiemstra, J.; Partilla, J.S.; Dersch, C.M.; Rothman, R.B.; et al. Herkinorin analogues with differential β-arrestin-2 interactions. J. Med. Chem. 2008, 51, 2421–2431. [Google Scholar] [CrossRef]
- Crowley, R.S.; Riley, A.P.; Sherwood, A.M.; Groer, C.E.; Shivaperumal, N.; Biscaia, M.; Paton, K.; Schneider, S.; Provasi, D.; Kivell, B.M.; et al. Synthetic Studies of Neoclerodane Diterpenes from Salvia divinorum: Identification of a Potent and Centrally Acting μ Opioid Analgesic with Reduced Abuse Liability. J. Med. Chem. 2016, 59, 11027–11038. [Google Scholar] [CrossRef]
- Holden, K.G.; Tidgewell, K.; Marquam, A.; Rothman, R.B.; Navarro, H.; Prisinzano, T.E. Synthetic studies of neoclerodane diterpenes from Salvia divinorum: Exploration of the 1-position. Bioorg. Med. Chem. Lett. 2007, 17, 6111–6115. [Google Scholar] [CrossRef]
- Béguin, C.; Richards, M.R.; Li, J.G.; Wang, Y.; Xu, W.; Liu-Chen, L.Y.; Carlezon, W.A., Jr.; Cohen, B.M. Synthesis and in vitro evaluation of salvinorin A analogues: Effect of configuration at C(2) and substitution at C(18). Bioorg. Med. Chem. Lett. 2006, 16, 4679–4685. [Google Scholar] [CrossRef]
- Lee, D.Y.; He, M.; Kondaveti, L.; Liu-Chen, L.Y.; Ma, Z.; Wang, Y.; Chen, Y.; Li, J.G.; Beguin, C.; Carlezon, W.A., Jr.; et al. Synthesis and in vitro pharmacological studies of C(4) modified salvinorin A analogues. Bioorg. Med. Chem. Lett. 2005, 15, 4169–4173. [Google Scholar] [CrossRef] [PubMed]
- Béguin, C.; Duncan, K.K.; Munro, T.A.; Ho, D.M.; Xu, W.; Liu-Chen, L.Y.; Carlezon, W.A., Jr.; Cohen, B.M. Modification of the furan ring of salvinorin A: Identification of a selective partial agonist at the kappa opioid receptor. Bioorg. Med. Chem. 2009, 17, 1370–1380. [Google Scholar] [CrossRef] [PubMed]
- Harding, W.W.; Schmidt, M.; Tidgewell, K.; Kannan, P.; Holden, K.G.; Dersch, C.M.; Rothman, R.B.; Prisinzano, T.E. Synthetic studies of neoclerodane diterpenes from Salvia divinorum: Selective modification of the furan ring. Bioorg. Med. Chem. Lett. 2006, 16, 3170–3174. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Xu, W.; Chen, F.; Liu-Chen, L.Y.; Ma, Z.; Lee, D.Y. Synthesis and biological evaluation of C-12 triazole and oxadiazole analogs of salvinorin A. Bioorg. Med. Chem. Lett. 2009, 19, 1301–1304. [Google Scholar] [CrossRef] [PubMed]
- Simpson, D.S.; Lovell, K.M.; Lozama, A.; Han, N.; Day, V.W.; Dersch, C.M.; Rothman, R.B.; Prisinzano, T.E. Synthetic studies of neoclerodane diterpenes from Salvia divinorum: Role of the furan in affinity for opioid receptors. Org. Biomol. Chem. 2009, 7, 3748–3756. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, T.; Kawamura, M.; Kikura-Hanajiri, R.; Takayama, H.; Goda, Y. The botanical origin of kratom (Mitragyna speciosa; Rubiaceae) available as abused drugs in the Japanese markets. J. Nat. Med. 2009, 63, 340–344. [Google Scholar] [CrossRef]
- Prozialeck, W.C.; Jivan, J.K.; Andurkar, S.V. Pharmacology of kratom: An emerging botanical agent with stimulant, analgesic and opioid-like effects. J. Am. Osteopath. Assoc. 2012, 112, 792–799. [Google Scholar]
- Takayama, H.; Ishikawa, H.; Kurihara, M.; Kitajima, M.; Aimi, N.; Ponglux, D.; Koyama, F.; Matsumoto, K.; Moriyama, T.; Yamamoto, L.T.; et al. Studies on the synthesis and opioid agonistic activities of mitragynine-related indole alkaloids: Discovery of opioid agonists structurally different from other opioid ligands. J. Med. Chem. 2002, 45, 1949–1956. [Google Scholar] [CrossRef]
- Kitajima, M.; Misawa, K.; Kogure, N.; Said, I.M.; Horie, S.; Hatori, Y.; Murayama, T.; Takayama, H. A new indole alkaloid, 7-hydroxyspeciociliatine, from the fruits of Malaysian Mitragyna speciose and its opioid agonistic activity. Nat. Med. 2006, 60, 28–35. [Google Scholar] [CrossRef]
- Kruegel, A.C.; Gassaway, M.M.; Kapoor, A.; Váradi, A.; Majumdar, S.; Filizola, M.; Javitch, J.A.; Sames, D. Synthetic and Receptor Signaling Explorations of the Mitragyna Alkaloids: Mitragynine as an Atypical Molecular Framework for Opioid Receptor Modulators. J. Am. Chem. Soc. 2016, 138, 6754–6764. [Google Scholar] [CrossRef]
- Raffa, R.B.; Beckett, J.R.; Brahmbhatt, V.N.; Ebinger, T.M.; Fabian, C.A.; Nixon, J.R.; Orlando, S.T.; Rana, C.A.; Tejani, A.H.; Tomazic, R.J. Orally active opioid compounds from a non-poppy source. J. Med. Chem. 2013, 56, 4840–4848. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, K.; Mizowaki, M.; Suchitra, T.; Takayama, H.; Sakai, S.; Aimi, N.; Watanabe, H. Antinociceptive action of mitragynine in mice: Evidence for the involvement of supraspinal opioid receptors. Life Sci. 1996, 59, 1149–1155. [Google Scholar] [CrossRef] [PubMed]
- Macko, E.; Weisbach, J.A.; Douglas, B. Some observations on the pharmacology of mitragynine. Arch. Int. Pharmacodyn. Ther. 1972, 198, 145–161. [Google Scholar] [PubMed]
- Thongpradichote, S.; Matsumoto, K.; Tohda, M.; Takayama, H.; Aimi, N.; Sakai, S.; Watanabe, H. Identification of opioid receptor subtypes in antinociceptive actions of supraspinally-administered mitragynine in mice. Life Sci. 1998, 62, 1371–1378. [Google Scholar] [CrossRef] [PubMed]
- Kumarnsit, E.; Keawpradub, N.; Nuankaew, W. Effect of Mitragyna speciosa aqueous extract on ethanol withdrawal symptoms in mice. Fitoterapia. 2007, 78, 182–185. [Google Scholar] [CrossRef]
- Shaik Mossadeq, W.M.; Sulaiman, M.R.; Tengku Mohamad, T.A.; Chiong, H.S.; Zakaria, Z.A.; Jabit, M.L.; Baharuldin, M.T.; Israf, D.A. Anti-inflammatory and antinociceptive effects of Mitragyna speciosa Korth methanolic extract. Med. Princ. Pract. 2009, 18, 378–384. [Google Scholar] [CrossRef]
- Utar, Z.; Majid, M.I.; Adenan, M.I.; Jamil, M.F.; Lan, T.M. Mitragynine inhibits the COX-2 mRNA expression and prostaglandin E2 production induced by lipopolysaccharide in RAW264.7 macrophage cells. J. Ethnopharmacol. 2011, 136, 75–82. [Google Scholar] [CrossRef]
- Zarembo, J.E.; Douglas, B.; Valenta, J.; Weisbach, J.A. Metabolites of mitragynine. J. Pharm. Sci. 1974, 63, 1407–1415. [Google Scholar] [CrossRef]
- Lounasmaa, M.; Tolvanen, A. The Corynantheine-Heteroyohimbine Group. Supplement to Part 4. Chapter 3. In Monoterpenoid Indole Alkaloids; Saxton, J.E., Ed.; John Wiley & Sons: Chichester, UK, 1994; pp. 57–159. [Google Scholar]
- Yamamoto, L.T.; Horie, S.; Takayama, H.; Aimi, N.; Sakai, S.; Yano, S.; Shan, J.; Pang, P.K.; Ponglux, D.; Watanabe, K. Opioid receptor agonistic characteristics of mitragynine pseudoindoxyl in comparison with mitragynine derived from Thai medicinal plant Mitragyna speciosa. Gen Pharmacol. 1999, 33, 73–81. [Google Scholar] [CrossRef]
- Takayama, H. Chemistry and pharmacology of analgesic indole alkaloids from the rubiaceous plant, Mitragyna speciosa. Chem. Pharm. Bull. 2004, 52, 916–928. [Google Scholar] [CrossRef]
- Matsumoto, K.; Hatori, Y.; Murayama, T.; Tashima, K.; Wongseripipatana, S.; Misawa, K.; Kitajima, M.; Takayama, H.; Horie, S. Involvement of μ-opioid receptors in antinociception and inhibition of gastrointestinal transit induced by 7-hydroxymitragynine, isolated from Thai herbal medicine Mitragyna speciosa. Eur. J. Pharmacol. 2006, 549, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, K.; Takayama, H.; Ishikawa, H.; Aimi, N.; Ponglux, D.; Watanabe, K.; Horie, S. Partial agonistic effect of 9-hydroxycorynantheidine on mu-opioid receptor in the guinea-pig ileum. Life Sci. 2006, 78, 2265–2271. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, K.; Narita, M.; Muramatsu, N.; Nakayama, T.; Misawa, K.; Kitajima, M.; Tashima, K.; Devi, L.A.; Suzuki, T.; Takayama, H.; et al. Orally active opioid μ/δ dual agonist MGM-16, a derivative of the indole alkaloid mitragynine, exhibits potent antiallodynic effect on neuropathic pain in mice. J. Pharmacol. Exp. Ther. 2014, 348, 383–392. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, K.; Takayama, H.; Narita, M.; Nakamura, A.; Suzuki, M.; Suzuki, T.; Murayama, T.; Wongseripipatana, S.; Misawa, K.; Kitajima, M.; et al. MGM-9 [(E)-methyl 2-(3-ethyl-7a,12a-(epoxyethanoxy)-9-fluoro-1,2,3,4,6,7,12,12b-octahydro-8-methoxyindolo[2,3-a]quinolizin-2-yl)-3-methoxyacrylate], a derivative of the indole alkaloid mitragynine: A novel dual-acting μ- and κ-opioid agonist with potent antinociceptive and weak rewarding effects in mice. Neuropharmacology 2008, 55, 154–165. [Google Scholar]
- Dhawan, B.N.; Cesselin, F.; Raghubir, R.; Reisine, T.; Bradley, P.B.; Portoghese, P.S.; Hamon, M. International Union of Pharmacology. XII. Classification of opioid receptors. Pharmacol. Rev. 1996, 48, 567–592. [Google Scholar]
- Váradi, A.; Marrone, G.F.; Palmer, T.C.; Narayan, A.; Szabó, M.R.; Le Rouzic, V.; Grinnell, S.G.; Subrath, J.J.; Warner, E.; Kalra, S.; et al. Mitragynine/Corynantheidine Pseudoindoxyls as Opioid Analgesics with Mu Agonism and Delta Antagonism, Which Do Not Recruit β-Arrestin-2. J. Med. Chem. 2016, 59, 8381–8397. [Google Scholar] [CrossRef]
- Chakraborty, S.; Majumdar, S. Natural Products for the Treatment of Pain: Chemistry and Pharmacology of Salvinorin A, Mitragynine, and Collybolide. Biochemistry 2021, 60, 1381–1400. [Google Scholar] [CrossRef]
- Mores, K.L.; Cummins, B.R.; Cassell, R.J.; van Rijn, R.M. A Review of the Therapeutic Potential of Recently Developed G Protein-Biased Kappa Agonists. Front. Pharmacol. 2019, 10, 407. [Google Scholar] [CrossRef]
- Kaserer, T.; Steinacher, T.; Kainhofer, R.; Erli, F.; Sturm, S.; Waltenberger, B.; Schuster, D.; Spetea, M. Identification and characterization of plant-derived alkaloids, corydine and corydaline, as novel mu opioid receptor agonists. Sci. Rep. 2020, 10, 13804. [Google Scholar] [CrossRef]
- Karimov, A.; Karimov, A.; Levkovich, M.G.; Abdullaev, N.D.; Shakirov, R. Berberis alkaloids. XXIII. Structure of turcherine. Chem. Nat. Comp. 1993, 29, 63–67. [Google Scholar] [CrossRef]
- Han, J.W.; Shim, S.H.; Jang, K.S.; Choi, Y.H.; Kim, H.; Choi, G.J. In vivo disease control efficacy of isoquinoline alkaloids isolated from Corydalis ternata against wheat leaf rust and pepper anthracnose. J. Microbiol. Biotechnol. 2018, 28, 262–266. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, A.; Zapata, M.; Sabando, C.; Bustamante, L.; von Baer, D.; Vergara, C.; Mardones, C. Flavonols, alkaloids, and antioxidant capacity of edible wild Berberis species from Patagonia. J. Agric. Food Chem. 2014, 62, 12407–12417. [Google Scholar] [CrossRef] [PubMed]
- Lei, F.; Yan, Z. Antinociceptive and Anti-inflammatory Effect of Corynoline in Different Nociceptive and Inflammatory Experimental Models. Appl. Biochem. Biotechnol. 2022, 194, 4783–4799. [Google Scholar] [CrossRef]
- Ding, B.; Zhou, T.; Fan, G.; Hong, Z.; Wu, Y. Qualitative and quantitative determination of ten alkaloids in traditional Chinese medicine Corydalis yanhusuo W.T. Wang by LC-MS/MS and LC-DAD. J. Agric. Food Chem. 2007, 45, 219–226. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.Z.; Xu, W.; Jensen, N.H.; Roth, B.L.; Liu-Chen, L.Y.; Lee, D.Y. Isoquinoline alkaloids isolated from Corydalis yanhusuo and their binding affinities at the dopamine D1 receptor. Molecules 2008, 13, 2303–2312. [Google Scholar] [CrossRef] [PubMed]
- Kupeli, E.; Kosar, M.; Yesilada, E.; Husnu, K.; Baser, C. A comparative study on the anti-inflammatory, anti-nociceptive and antipyretic effects of isoquinoline alkaloids from the roots of Turkish Berberis species. Life Sci. 2002, 72, 645–657. [Google Scholar] [CrossRef]
- Nishiyama, Y.; Moriyasu, M.; Ichimaru, M.; Iwasa, K.; Kato, A.; Mathenge, S.G.; Chalo Mutiso, P.B.; Juma, F.D. Antinociceptive effects of the extracts of Xylopia parviflora bark and its alkaloidal components in experimental animals. J. Nat. Med. 2010, 64, 9–15. [Google Scholar] [CrossRef]
- Wang, L.; Zhang, Y.; Wang, Z.; Gong, N.; Kweon, T.D.; Vo, B.; Wang, C.; Zhang, X.; Chung, J.Y.; Alachkar, A.; et al. The Antinociceptive Properties of the Corydalis yanhusuo Extract. PLoS ONE 2016, 11, e0162875. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, C.; Wang, L.; Parks, G.S.; Zhang, X.; Guo, Z.; Ke, Y.; Li, K.W.; Kim, M.K.; Vo, B.; et al. A novel analgesic isolated from a traditional Chinese medicine. Curr. Biol. 2014, 24, 117–123. [Google Scholar] [CrossRef]
- Xu, Y.; Sun, J.; Li, W.; Zhang, S.; Yang, L.; Teng, Y.; Lv, K.; Liu, Y.; Su, Y.; Zhang, J.; et al. Analgesic effect of the main components of Corydalis yanhusuo (yanhusuo in Chinese) is caused by inhibition of voltage gated sodium channels. J. Ethnopharmacol. 2021, 280, 114457. [Google Scholar] [CrossRef]
- Hsu, B.; Kin, K.C. Pharmacological study of tetrahydropalmatine and its analogs. A new type of central depressants. Arch. Int. Pharmacodyn. Ther. 1962, 139, 318–327. [Google Scholar] [PubMed]
- Hu, J.Y.; Jin, G.Z. Supraspinal D2 receptor involved in antinociception induced by l-tetrahydropalmatine. Zhong Guo Yao Li Xue Bao 1999, 20, 715–719. [Google Scholar]
- Guo, Z.; Man, Y.; Wang, X.; Jin, H.; Sun, X.; Su, X.; Hao, J.; Mi, W. Levo-tetrahydropalmatine attenuates oxaliplatin-induced mechanical hyperalgesia in mice. Sci. Rep. 2014, 4, 3905. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.H.; Wu, D.L.; Gao, L.Y.; Fang, Y.; Ge, W.H. L-Tetrahydropalmatine alleviates mechanical hyperalgesia in models of chronic inflammatory and neuropathic pain in mice. NeuroReport 2016, 27, 476–480. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, R.R.; Sun, W.; Lou, C.; Yang, F.; He, T.; Wang, X.L.; Cao, F.L.; Chen, J. Analgesic effect of dl-THP on inflammatory pain mediated by suppressing spinal TRPV1 and P2X3 receptors in rats. Front. Biosci. Landmark 2021, 26, 1–10. [Google Scholar] [CrossRef]
- Kang, D.W.; Moon, J.Y.; Choi, J.G.; Kang, S.Y.; Ryu, Y.; Park, J.B.; Lee, J.H.; Kim, H.W. Antinociceptive Profile of Levo-tetrahydropalmatine in Acute and Chronic Pain Mice Models: Role of spinal sigma-1 receptor. Sci. Rep. 2016, 6, 37850. [Google Scholar] [CrossRef]
- Liu, J.; Dai, R.; Damiescu, R.; Efferth, T.; Lee, D.Y.W. Role of Levo-tetrahydropalmatine and its metabolites for management of chronic pain and opioid use disorders. Phytomedicine 2021, 90, 153594. [Google Scholar] [CrossRef]
- Zhou, L.; Ao, L.; Yan, Y.; Li, C.; Li, W.; Ye, A.; Liu, J.; Hu, Y.; Fang, W.; Li, Y. Levo-corydalmine Attenuates Vincristine-Induced Neuropathic Pain in Mice by Upregulating the Nrf2/HO-1/CO Pathway to Inhibit Connexin 43 Expression. Neurotherapeutics 2020, 17, 340–355. [Google Scholar] [CrossRef]
- Muhammad, N.; Shrestha, R.L.; Adhikari, A.; Wadood, A.; Khan, H.; Khan, A.Z.; Maione, F.; Mascolo, N.; De Feo, V. First evidence of the analgesic activity of govaniadine, an alkaloid isolated from Corydalis govaniana Wall. Nat. Prod. Res. 2015, 29, 430–437. [Google Scholar] [CrossRef]
- Oliveira de Veras, B.; Melo de Oliveira, M.B.; Granja da Silva Oliveira, F.; Queiroz Dos Santos, Y.; Saturnino de Oliveira, J.R.; Lúcia de Menezes Lima, V.; Guedes da Silva Almeida, J.R.; Maria do Amaral Ferraz Navarro, D.; Ribeiro de Oliveira Farias de Aguiar, J.C.; Aguiar, J.D.S.; et al. Chemical composition and evaluation of the antinociceptive, antioxidant and antimicrobial effects of essential oil from Hymenaea cangaceira (Pinto, Mansano & Azevedo) native to Brazil: A natural medicine. J. Ethnopharmacol. 2020, 247, 112265. [Google Scholar]
- Oliveira, F.G.; de Lima-Saraiva, S.R.; Oliveira, A.P.; Rabêlo, S.V.; Rolim, L.A.; Almeida, J.R. Influence of the Extractive Method on the Recovery of Phenolic Compounds in Different Parts of Hymenaea martiana Hayne. Pharmacogn. Res. 2016, 8, 270–275. [Google Scholar]
- Magalhães, J.F.G.; Viana, C.F.G.; Aragão, A.G.M.; Moraes, V.G.; Ribeiro, R.A.; Vale, M.R. Analgesic and antiinflammatory activities of Ageratum conyzoides in rats. Phytoth. Res. 1997, 11, 183–188. [Google Scholar] [CrossRef]
- Manthey, J.A.; Bendele, P. Anti-inflammatory activity of an orange peel polymethoxylated flavone, 3′,4′,3,5,6,7,8-heptamethoxyflavone, in the rat carrageenan/paw edema and mouse lipopolysaccharide-challenge assays. J. Agric. Food Chem. 2008, 56, 9399–9403. [Google Scholar] [CrossRef] [PubMed]
- Faqueti, L.G.; Brieudes, V.; Halabalaki, M.; Skaltsounis, A.L.; Nascimento, L.F.; Barros, W.M.; Santos, A.R.; Biavatti, M.W. Antinociceptive and anti-inflammatory activities of standardized extract of polymethoxyflavones from Ageratum conyzoides. J. Ethnopharmacol. 2016, 194, 369–377. [Google Scholar] [CrossRef]
- Kim, M.J.; Lee, H.H.; Jeong, J.W.; Seo, M.J.; Kang, B.W.; Park, J.U.; Kim, K.S.; Cho, Y.S.; Seo, K.I.; Kim, G.Y.; et al. Anti-inflammatory effects of 5-hydroxy-3,6,7,8,3′,4′-hexamethoxyflavone via NF-κB inactivation in lipopolysaccharide-stimulated RAW 264.7 macrophage. Mol. Med. Rep. 2014, 9, 1197–1203. [Google Scholar] [CrossRef] [PubMed]
- Sukmawan, Y.P.; Anggadiredja, K.; Adnyana, I.K. Anti-Neuropathic Pain Activity of Ageratum conyzoides L due to the Essential Oil Components. CNS Neurol. Disord. Drug Targets 2021, 20, 181–189. [Google Scholar] [CrossRef]
- Gurib-Fakim, A. Medicinal plants: Traditions of yesterday and drugs of tomorrow. Mol. Asp. Med. 2006, 27, 1–93. [Google Scholar] [CrossRef]
- Rodrigues, E.; Mendes, F.R.; Negri, G. Plants indicated by Brazilian Indians to central nervous system disturbances: A bibliographical approach. Curr. Med. Chem. 2006, 6, 211–244. [Google Scholar] [CrossRef]
- do Nascimento, J.R.; de Jesus Alves Miranda, A.; Vieira, F.C.; Rodrigues, C.D.P.; Vasconcelos, L.N.; Filho, J.L.P.; Lopes, A.C.C.B.; Tangerina, M.M.P.; Vilegas, W.; da Rocha, C.Q. A Review of the Phytochemistry and Pharmacological Properties of the Genus Arrabidaea. Pharmaceuticals 2022, 15, 658. [Google Scholar] [CrossRef]
- Novaes, P.; Molinillo, J.M.G.; Varela, R.M.; Macías, F.A. Ecological phytochemistry of Cerrado (Brazilian savanna) plants. Phytochem. Rev. 2013, 12, 839–855. [Google Scholar] [CrossRef]
- Da Rocha, C.Q.; Vilela, F.C.; Cavalcante, G.P.; Santa-Cecília, F.V.; Santos-e-Silva, L.; dos Santos, M.H.; Giusti-Paiva, A. Anti-inflammatory and antinociceptive effects of Arrabidaea brachypoda (DC.) Bureau roots. J. Ethnopharmacol. 2011, 133, 396–401. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, V.P.; Rocha, C.Q.D.; Périco, L.L.; Santos, R.C.D.; Ohara, R.; Nishijima, C.M.; Ferreira Queiroz, E.; Wolfender, J.L.; Rocha, L.R.M.D.; Santos, A.R.S.; et al. Involvement of Opioid System, TRPM8, and ASIC Receptors in Antinociceptive Effect of Arrabidaea brachypoda (DC) Bureau. Int. J. Mol. Sci. 2017, 18, 2304. [Google Scholar] [CrossRef] [PubMed]
- Da Rocha, C.Q.; Queiroz, E.F.; Meira, C.S.; Moreira, D.R.M.; Soares, M.B.P.; Marcourt, L.; Vilegas, W.; Wolfender, J.-L. Dimeric Flavonoids from Arrabidaea brachypoda and Assessment of Their Anti-Trypanosoma cruzi Activity. J. Nat. Prod. 2014, 77, 1345–1350. [Google Scholar] [CrossRef] [PubMed]
- Salgado, C.; Morin, H.; Coriolano de Aquino, N.; Neff, L.; Quintino da Rocha, C.; Vilegas, W.; Marcourt, L.; Wolfender, J.L.; Jordan, O.; Ferreira Queiroz, E.; et al. In Vitro Anti-Inflammatory Activity in Arthritic Synoviocytes of A. brachypoda Root Extracts and Its Unusual Dimeric Flavonoids. Molecules 2020, 25, 5219. [Google Scholar] [CrossRef] [PubMed]
- Duke, J.A.; Bogenschutz-Godwin, M.J.; duCellier, J.; Duke, A.K. Hand Book of Medical Herbs 2002, 2nd ed.; CRC Press: Boca Raton, FL, USA, 2002; 473p. [Google Scholar]
- Nakamura, S.; Nakashima, S.; Tanabe, G.; Oda, Y.; Yokota, N.; Fujimoto, K.; Matsumoto, T.; Sakuma, R.; Ohta, T.; Ogawa, K.; et al. Alkaloid constituents from flower buds and leaves of sacred lotus (Nelumbo nucifera, Nymphaeaceae) with melanogenesis inhibitory activity in B16 melanoma cells. Bioorg. Med. Chem. 2013, 21, 779–787. [Google Scholar] [CrossRef]
- Saengkhae, C.; Arunnopparat, W.; Sungkhajorn, P. Antioxidant activity of Nelumbo nucifera Gaertn on oxidative stress-induced Erythrocyte hemolysis in hypertensive and normotensive rats. Thai J. Physiol. Sci. 2008, 20, 70–78. [Google Scholar]
- Mathew, M.; Subramanian, S. In vitro screening for anticholinesterase and antioxidant activity of methanolic extracts of Ayurvedic medicinal plants used for cognitive disorders. PLoS ONE 2014, 9, e86804. [Google Scholar] [CrossRef]
- Tanahashi, T.; Yamada, J.; Nakajima, H.; Sun, S.-J. Method and Health Food for Preventing and/or Alleviating Psychiatric Disorder, and/or for Effectuating Sedation. U.S. Patent US20060030586A1, 21 September 2010. [Google Scholar]
- Nakajima, H.; Sugimoto, Y.; Sun, S.-J.; Tanahashi, T.; Yamada, J. Benzylisoquinoline Derivative- or Bisbenzylisoquinoline Derivative-Containing Psychotropic Agent, Analgesic and/or Antiphlogistic, and Health Food. U.S. Patent US20070027181A1, 1 February 2007. [Google Scholar]
- O’Mahony Carey, S. Psychoactive Substances: A Guide to Ethnobotanical Plants and Herbs, Synthetic Chemicals, Compounds and Products. 2010. Available online: http://www.drugs.ie/resourcesfiles/guides/Psychoactive_substances_low_res.pdf (accessed on 23 September 2022).
- Kumarihamy, M.; León, F.; Pettaway, S.; Wilson, L.; Lambert, J.A.; Wang, M.; Hill, C.; McCurdy, C.R.; ElSohly, M.A.; Cutler, S.J.; et al. In vitro opioid receptor affinity and in vivo behavioral studies of Nelumbo nucifera flower. J. Ethnopharmacol. 2015, 174, 57–65. [Google Scholar] [CrossRef]
- Trevisan, G.; Rossato, M.F.; Tonello, R.; Hoffmeister, C.; Klafke, J.Z.; Rosa, F.; Pinheiro, K.V.; Pinheiro, F.V.; Boligon, A.A.; Athayde, M.L.; et al. Gallic acid functions as a TRPA1 antagonist with relevant antinociceptive and antiedematogenic effects in mice. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2014, 387, 679–689. [Google Scholar] [CrossRef]
- Mehrotra, A.; Shanbhag, R.; Chamallamudi, M.R.; Singh, V.P.; Mudgal, J. Ameliorative effect of caffeic acid against inflammatory pain in rodents. Eur. J. Pharmacol. 2011, 666, 80–86. [Google Scholar] [CrossRef]
- Lv, W.-H.; Zhang, L.; Wu, S.-J.; Chen, S.-Z.; Zhu, X.-B.; Pan, J.-C. Analgesic effect of ferulic acid on CCI mice: Behavior and neurobiological analysis. Zhongguo Zhong Yao Za Zhi 2013, 38, 3736–3741. [Google Scholar]
- Borghi, S.M.; Carvalho, T.T.; Staurengo-Ferrarietal, L. Vitexin inhibits inflammatory pain in mice by targeting TRPV1, oxidative stress, and cytokines. J. Nat. Prod. 2013, 76, 1141–1149. [Google Scholar] [CrossRef] [PubMed]
- Pinheiro, M.M.G.; Boylan, F.; Fernandes, P.D. Antinociceptive effect of the Orbignya speciosa Mart. (Babassu) leaves: Evidence for the involvement of apigenin. Life Sci. 2012, 91, 293–300. [Google Scholar] [CrossRef] [PubMed]
- Abdul Rahim, M.H.; Zakaria, Z.A.; Mohd Sani, M.H.; Omar, M.H.; Yakob, Y.; Cheema, M.S.; Ching, S.M.; Ahmad, Z.; Abdul Kadir, A. Methanolic Extract of Clinacanthus nutans Exerts Antinociceptive Activity via the Opioid/Nitric Oxide-Mediated, but cGMP-Independent, Pathways. Evid.-Based Complement. Altern. Med. 2016, 2016, 1494981. [Google Scholar] [CrossRef]
- Zakaria, Z.A.; Abdul Rahim, M.H.; Roosli, R.A.J.; Mohd Sani, M.H.; Marmaya, N.H.; Omar, M.H.; The, L.K.; Salleh, M.Z. Antinociceptive Activity of Petroleum Ether Fraction of Clinacanthus nutans Leaves Methanolic Extract: Roles of Nonopioid Pain Modulatory Systems and Potassium Channels. BioMed Res. Int. 2019, 2019, 6593125. [Google Scholar] [CrossRef]
- Zakaria, Z.A.; Abdul Rahim, M.H.; Roosli, R.A.J.; Mohd Sani, M.H.; Omar, M.H.; Mohd Tohid, S.F.; Othman, F.; Ching, S.M.; Abdul Kadir, A. Antinociceptive Activity of Methanolic Extract of Clinacanthus nutans Leaves: Possible Mechanisms of Action Involved. Pain Res. Manag. 2018, 2018, 9536406. [Google Scholar] [CrossRef] [PubMed]
- Dasgupta, T.; Banerjee, S.; Yadava, P.K.; Rao, A.R. Chemopreventive potential of Azadirachta indica (Neem) leaf extract in murine carcinogenesis model systems. J. Ethnopharmacol. 2004, 92, 23–36. [Google Scholar] [CrossRef]
- Elumalai, P.; Gunadharini, D.N.; Senthilkumar, K.; Banudevi, S.; Arunkumar, R.; Benson, C.S.; Sharmila, G.; Arunakaran, J. Ethanolic neem (Azadirachta indica A. Juss) leaf extract induces apoptosis and inhibits the IGF signaling pathway in breast cancer cell lines. Biomed. Prev. Nutr. 2012, 2, 59–68. [Google Scholar] [CrossRef]
- Botez, S.A.; Herrmann, D.N. Sensory neuropathies, from symptoms to treatment. Curr. Opin. Neurol. 2010, 23, 502–508. [Google Scholar] [CrossRef]
- Raji, Y.; Ogunwande, I.A.; Osadebe, C.A.; John, G. Effects of Azadirachta indica extract on gastric ulceration and acid secretion in rats. J. Ethnopharmacol. 2004, 90, 167–170. [Google Scholar] [CrossRef]
- Chattopadhyay, R.; Bandyopadhyay, M. Effect of Aza dirachta indica leaf extract on serum lipid profile changes in normal and streptozotocin induced diabetic rats. Afr. J. Biomed. Res. 2005, 8, 101–104. [Google Scholar]
- Chattopadhyay, R.R. Possible mechanism of antihyper-glycemic effect of Azadirachta indica leaf extract. Part IV. Gen. Pharmacol. 1996, 27, 431–434. [Google Scholar] [CrossRef] [PubMed]
- Kanagasanthosh, K.; Kavirajan, V.; Shanmugapriyan, S. Evaluation of antinociceptive activity of ethanolic extract of Azadirachta indica leaves. Int. J. Res. Dev. Pharm. Life Sci. 2016, 5, 1934–1940. [Google Scholar]
- Batista, F.L.A.; Lima, L.M.G.; Abrante, I.A.; de Araújo, J.I.F.; Batista, F.L.A.; Abrante, I.A.; Magalhães, E.A.; de Lima, D.R.; Lima, M.D.C.L.; do Prado, B.S.; et al. Antinociceptive activity of ethanolic extract of Azadirachta indica A. Juss (Neem, Meliaceae) fruit through opioid, glutamatergic and acid-sensitive ion pathways in adult zebrafish (Danio rerio). Biomed. Pharmacother. 2018, 108, 408–416. [Google Scholar] [CrossRef]
- Kandhare, A.D.; Mukherjee, A.A.; Bodhankar, S.L. Neuroprotective effect of Azadirachta indica standardized extract in partial sciatic nerve injury in rats: Evidence from anti-inflammatory, antioxidant and anti-apoptotic studies. EXCLI J. 2017, 16, 546–565. [Google Scholar]
- Koriem, K.M. Review on pharmacological and toxicologyical effects of oleum azadirachti oil. Asian Pac. J. Trop. Biomed. 2013, 3, 834–840. [Google Scholar] [CrossRef]
- Pillai, N.R.; Santhakumari, G. Anti-arthritic and anti-inflammatory actions of nimbidin. Planta Med. 1981, 43, 59–63. [Google Scholar] [CrossRef] [PubMed]
- Shankaranarayan, D. Effect of neem oil and its constituents on cotton pellet inflammation. Mediscope 1978, 20, 273–274. [Google Scholar]
- Soares, D.G.; Godin, A.M.; Menezes, R.R.; Nogueira, R.D.; Brito, A.M.; Melo, I.S.; Coura, G.M.; Souza, D.G.; Amaral, F.A.; Paulino, T.P.; et al. Anti-inflammatory and antinociceptive activities of azadirachtin in mice. Planta Med. 2014, 80, 630–636. [Google Scholar] [CrossRef]
- Hoff, R.; Daguer, H.; Deolindo, C.T.P.; de Melo, A.P.Z.; Durigon, J. Phenolic compounds profile and main nutrients parameters of two underestimated non-conventional edible plants: Pereskia aculeata Mill. (ora-pro-nóbis) and Vitex megapotamica (Spreng.) Moldenke (tarumã) fruits. Food Res. Int. 2022, 162, 112042. [Google Scholar] [CrossRef]
- Hamann, F.R.; Zago, A.M.; Rossato, M.F.; Beck, V.R.; Mello, C.F.; de Brum, T.F.; de Carvalho, L.M.; Faccin, H.; Oliveira, S.M.; Rubin, M.A. Antinociceptive and antidepressant-like effects of the crude extract of Vitex megapotamica in rats. J. Ethnopharmacol. 2016, 192, 210–216. [Google Scholar] [CrossRef] [PubMed]
- Cai, S.; Bellampalli, S.S.; Yu, J.; Li, W.; Ji, Y.; Wijeratne, E.M.K.; Dorame, A.; Luo, S.; Shan, Z.; Khanna, M.; et al. (−)-Hardwickiic Acid and Hautriwaic Acid Induce Antinociception via Blockade of Tetrodotoxin-Sensitive Voltage-Dependent Sodium Channels. ACS Chem. Neurosci. 2019, 10, 1716–1728. [Google Scholar] [CrossRef]
- Pittaluga, A.; Olivero, G.; Di Prisco, S.; Merega, E.; Bisio, A.; Romussi, G.; Grilli, M.; Marchi, M. Effects of the neoclerodane Hardwickiic acid on the presynaptic opioid receptors which modulate noradrenaline and dopamine release in mouse central nervous system. Neurochem. Int. 2013, 62, 354–359. [Google Scholar] [CrossRef]
- Símaro, G.V.; Lemos, M.; Mangabeira da Silva, J.J.; Ribeiro, V.P.; Arruda, C.; Schneider, A.H.; Wagner de Souza Wanderley, C.; Carneiro, L.J.; Mariano, R.L.; Ambrósio, S.R.; et al. Antinociceptive and anti-inflammatory activities of Copaifera pubiflora Benth oleoresin and its major metabolite ent-hardwickiic acid. J. Ethnopharmacol. 2021, 271, 113883. [Google Scholar] [CrossRef] [PubMed]
- Proenca, C.E.B.; Nic-Lughadha, E.M.; Lucas, E.; Woodgyer, E.M. Algrizea (Myrteae, Myrtaceae): A new genus from the Highlands of Brazil. Syst. Bot. 2006, 31, 320–326. [Google Scholar] [CrossRef]
- de Veras, B.O.; Dos Santos, Y.Q.; Oliveira, F.G.D.S.; Almeida, J.R.G.D.S.; da Silva, A.G.; Correia, M.T.D.S.; Diniz, K.M.; de Oliveira, J.R.S.; Lima, V.L.M.; Navarro, D.M.D.A.F.; et al. Algrizea Minor Sobral, Faria & Proenca (Myrteae, Myrtaceae): Chemical composition, antinociceptive, antimicrobial and antioxidant activity of essential oil. Nat Prod. Res. 2020, 34, 3013–3017. [Google Scholar] [CrossRef]
- Amorim, A.C.L.; Lima, C.K.F.; Hovell, A.M.C.; Miranda, A.L.P.; Rezende, C.M. Antinociceptive and hypothermic evaluation of the leaf essential oil and isolated terpenoids from Eugenia uniflora L. (Brazilian Pitanga). Phytomedicine 2009, 16, 923–928. [Google Scholar] [CrossRef]
- Küpeli, E.; Sahin, F.P.; Yeşilada, E.; Caliş, I.; Ezer, N. In vivo anti-inflammatory and antinociceptive activity evaluation of phenolic compounds from Sideritis stricta. Z. Naturforsch. C J. Biosci. 2007, 62, 519–525. [Google Scholar] [CrossRef]
- Isacchi, B.; Iacopi, R.; Bergonzi, M.C.; Ghelardini, C.; Galeotti, N.; Norcini, M.; Vivoli, E.; Vincieri, F.F.; Bilia, A.R. Antihyperalgesic activity of verbascoside in two models of neuropathic pain. J. Pharm. Pharmacol. 2011, 63, 594–601. [Google Scholar] [CrossRef]
- Amin, B.; Poureshagh, E.; Hosseinzadeh, H. The effect of verbascoside in neuropathic pain induced by chronic constriciton injury in rats. Phytother. Res. 2016, 30, 128–135. [Google Scholar] [CrossRef]
- Daels-Rakotoarison, D.A.; Seidel, V.; Gressier, B.; Brunet, C.; Tillequin, F.; Bailleul, F.; Luyckx, M.; Dine, T.; Cazin, M.; Cazin, J.C. Neurosedative and antioxidant activities of phenylpropanoids from Ballota nigra. Arzneimittelforschung 2000, 50, 16–23. [Google Scholar] [CrossRef] [PubMed]
- Isacchi, B.; Bergonzi, M.C.; Iacopi, R.; Ghelardini, C.; Galeotti, N.; Bilia, A.R. Liposomal Formulatin to increase stability and prolong antineuropathic activity of verbascoside. Planta Med. 2016, 83, 412–419. [Google Scholar] [PubMed]
- Seelinger, G.; Merfort, I.; Schempp, C. Anti-oxidant, anti-inflammatory and anti-allergic activities of luteolin. Planta Med. 2008, 74, 1667–1677. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Li, D.; Zong, Y.; Zhu, H.; Pan, D.; Xu, T.; Wang, T. Luteolin inhibits inflammatory responses via p/38/MK2/TTP-mediated mRNA stability. Molecules 2013, 18, 8083–8094. [Google Scholar] [CrossRef] [PubMed]
- Oeckinghaus, A.; Ghosh, S. The NF-kB family of transcription factors and its regulation. Cold Spring Harb. Perspect. 2009, 1, a000034. [Google Scholar]
- Hara, K.; Haranishi, Y.; Terada, T.; Takahashi, Y.; Nakamura, M.; Sata, T. Effects of intrathecal and intracerebroventricular administration of luteolin in a rat neuropathic pain model. Pharmacol. Biochem. Behav. 2014, 125, 78–84. [Google Scholar] [CrossRef]
- Jung, H.J. Anti-inflammatory activity of Korean thistle Cirsium maackii and its major flavonoid, luteolin 5-O-glucoside. Food Chem. Toxicol. 2012, 50, 2171–2179. [Google Scholar] [CrossRef]
- Jin, M.S. Luteolin-7-O-glucoside suppresses leukotriene C(4) production and degranulation by inhibiting the phosphorylation of mitogen activated protein kinases and phospholipase Cgamma1 in activated mouse bone marrow-derived mast cells. Biol. Pharm. Bull. 2011, 34, 1032–1036. [Google Scholar] [CrossRef]
- Mesia-Vela, S.; Souccar, C.; Lima-Landman, M.T.; Lapa, A.J. Pharmacological study of Stachytarpheta cayennensis Vahl in rodents. Phytomedicine 2004, 11, 616–624. [Google Scholar] [CrossRef]
- Schapoval, E.E.; Vargas, M.R.; Chaves, C.G.; Bridi, R.; Zuanazzi, J.A.; Henriques, A.T. Antiinflammatory and antinociceptive activities of extracts and isolated compounds from Stachytarpheta cayennensis. J. Ethnopharmacol. 1998, 60, 53–59. [Google Scholar] [CrossRef]
- Szpakowska, M.; Decker, A.M.; Meyrath, M.; Palmer, C.B.; Blough, B.E.; Namjoshi, O.A.; Chevigné, A. The natural analgesic conolidine targets the newly identified opioid scavenger ACKR3/CXCR7. Signal Transduct. Target. Ther. 2021, 6, 209. [Google Scholar] [CrossRef] [PubMed]
- Edinoff, A.N.; Patel, A.S.; Baker, M.W.; Lawson, J.; Wolcott, C.; Cornett, E.M.; Sadegi, K.; Kaye, A.M.; Kaye, A.D. Conolidine: A Novel Plant Extract for Chronic Pain. Anesth. Pain Med. 2021, 11, e121438. [Google Scholar] [CrossRef] [PubMed]
- Arita, T.; Asano, M.; Kubota, K.; Domon, Y.; Machinaga, N.; Shimada, K. Discovery of conolidine derivative DS39201083 as a potent novel analgesic without mu opioid agonist activity. Bioorg. Med. Chem. Lett. 2019, 29, 1938–1942. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Yunden, J.; Sonoda, S.; Doyama, N.; Lipkowski, A.W.; Kawamura, Y.; Yoshikawa, M. Rubiscolin, a δ selective opioid peptide derived from plant Rubisco. FEBS Lett. 2001, 509, 213–217. [Google Scholar] [CrossRef]
- Cassell, R.J.; Mores, K.L.; Zerfas, B.L.; Mahmoud, A.H.; Lill, M.A.; Trader, D.J.; van Rijn, R.M. Rubiscolins are naturally occurring G protein-biased delta opioid receptor peptides. Eur. Neuropsychopharmacol. 2019, 29, 450–456. [Google Scholar] [CrossRef]
- Yang, S.; Sonoda, S.; Chen, L.; Yoshikawa, M. Structure–activity relationship of rubiscolins as δ opioid peptides. Peptides 2003, 24, 503–508. [Google Scholar] [CrossRef]
- Jorge, R.; Leite, J.; Oliveira, A.; Tagliati, C. Evaluation of anti-nociceptive, anti-inflammatory and antiulcerogenic activities of Maytenus ilicifolia. J. Ethnopharmacol. 2004, 94, 93–100. [Google Scholar] [CrossRef]
- Sosa, S.; Morelli, C.; Tubaro, A.; Cairoli, P.; Speranza, G.; Manitto, P. Anti-inflammatory activity of Maytenus senegalensis root extracts and of maytenoic acid. Phytomedicine 2007, 14, 109–114. [Google Scholar] [CrossRef]
- Veloso, C. deC.; Rodrigues, V.G.; Ferreira, R.C.; Duarte, L.P.; Klein, A.; Duarte, I.D.; Romero, T.R.; Perez, A.d.C. TIngenone, a Pentacyclic Triterpene, Induces Peripheral Antinociception Due to Opioidergic Activation. Planta Med. 2014, 80, 1615–1621. [Google Scholar]
- Veloso, C.C.; Ferreira, R.C.M.; Rodrigues, V.G.; Duarte, L.P.; Klein, A.; Duarte, I.D.; Romero, T.R.L.; Perez, A.C. Tingenone, a pentacyclic triterpene, induces peripheral antinociception due to cannabinoid receptors activation in mice. Inflammopharmacology 2017, 26, 227–233. [Google Scholar] [CrossRef]
- de Carvalho Veloso, C.; Rodrigues, V.G.; Ferreira, R.C.; Duarte, L.P.; Klein, A.; Duarte, I.D.; Romero, T.R.; de Castro Perez, A. Tingenone, a pentacyclic triterpene, induces peripheral antinociception due to NO/cGMP and ATP-sensitive K+ channels pathway activation in mice. Eur. J. Pharmacol. 2015, 755, 1–5. [Google Scholar] [CrossRef]
- Radulović, N.S.; Miltojević, A.B.; McDermott, M.; Waldren, S.; Parnell, J.A.; Pinheiro, M.M.; Fernandes, P.D.; de Sousa Menezes, F. Identification of a new antinociceptive alkaloid isopropyl N-methylanthranilate from the essential oil of Choisya ternata Kunth. J. Etnopharmacol. 2011, 135, 610–619. [Google Scholar] [CrossRef]
- Fairbanks, C.; Stone, L.; Kitto, K.; Nguyen, H.E. α2C-adrenergic receptors mediate spinal analgesia and adrenergic-opioid synergy. J. Pharmacol. Exp. Ther. 2002, 300, 282–290. [Google Scholar] [CrossRef] [PubMed]
- Stein, C.; Clark, J.D.; Oh, U.; Vasko, M.R.; Wilcox, G.L.; Overland, A.C.; Vanderah, T.W.; Spencer, R.H. Peripheral mechanism of pain and analgesia. Brain Res. Rev. 2008, 60, 90–113. [Google Scholar] [CrossRef] [PubMed]
- Martins Gomes Pinheiro, M.; Miltojević, A.; Niko, S.; Radulović Abdul-Wahab, I.; Boylan, F.; Dias Fernandes, P. Anti-Inflammatory Activity of Choisya ternata Kunth Essential Oil, Ternanthranin, and Its Two Synthetic Analogs (Methyl and Propyl N-Methylanthranilates). PLoS ONE 2015, 10, e0121063. [Google Scholar]
- Valle-Dorado, M.G.; Hernández-León, A.; Nani-Vázquez, A.; Ángeles-López, G.E.; González-Trujano, M.E.; Ventura-Martínez, R. Antinociceptive effect of Mansoa alliacea polar extracts involves opioid receptors and nitric oxide in experimental nociception in mice. Biomed. Pharmacother. 2022, 152, 113253. [Google Scholar] [CrossRef]
- Hamann, F.R.; Brusco, I.; de Campos Severo, G.; de Carvalho, L.M.; Faccin, H.; Gobo, L.; Oliveira, S.M.; Rubin, M.A. Mansoa alliacea extract presents antinociceptive effect in a chronic inflammatory pain model in mice through opioid mechanisms. Neurochem. Int. 2019, 122, 157–169. [Google Scholar] [CrossRef] [PubMed]
- Pascoal, A.C.; Ehrenfried, C.A.; Lopez, B.G.; de Araujo, T.M.; Pascoal, V.D.; Gilioli, R.; Anhe, G.F.; Ruiz, A.L.; Carvalho, J.E.; Stefanello, M.E. Antiproliferative Activity and Induction of Apoptosis in PC-3 Cells by the Chalcone Cardamonin from Campomanesia adamantium (Myrtaceae) in a Bioactivity-Guided Study. Molecules 2014, 19, 1843. [Google Scholar] [CrossRef] [PubMed]
- Qin, Y.; Sun, C.-Y.; Lu, F.-R.; Shu, X.-R.; Yang, D.; Chen, L.; She, X.-M.; Gregg, N.M.; Guo, T.; Hu, Y. Cardamonin exerts potent activity against multiple myeloma through blockade of NF-κB pathway in vitro. Leuk. Res. 2012, 36, 514–520. [Google Scholar] [CrossRef]
- Cho, M.; Ryu, M.; Jeong, Y.; Chung, Y.-H.; Kim, D.-E.; Cho, H.-S.; Kang, S.; Han, J.-S.; Chang, M.-Y.; Lee, C.-K.; et al. Cardamonin suppresses melanogenesis by inhibition of Wnt/β-catenin signaling. Biochem. Biophys. Res. Commun. 2009, 390, 500–505. [Google Scholar] [CrossRef]
- Tang, Y.; Fang, Q.; Shi, D.; Niu, P.; Chen, Y.; Deng, J. mTOR inhibition of cardamonin on antiproliferation of A549 cells is involved in a FKBP12 independent fashion. Life Sci. 2014, 99, 44–51. [Google Scholar] [CrossRef] [PubMed]
- Wei, Z.; Yang, J.; Xia, Y.F.; Huang, W.Z.; Wang, Z.T.; Dai, Y. Cardamonin protects septic mice from acute lung injury by preventing endothelial barrier dysfunction. J. Biochem. Mol. Toxicol. 2012, 26, 282–290. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.-T.; Lau, C.-W.; Chan, F.L.; Yao, X.; Chen, Z.-Y.; He, Z.-D.; Huang, Y. Vasorelaxant effects of cardamonin and alpinetin from Alpinia henryi K. Schum. J. Cardiovasc. Pharmacol. 2001, 37, 596–606. [Google Scholar] [CrossRef]
- El-Naga, R.N. Pre-treatment with cardamonin protects against cisplatin-induced nephrotoxicity in rats: Impact on NOX-1, inflammation and apoptosis. Toxicol. Appl. Pharmacol. 2014, 274, 87–95. [Google Scholar] [CrossRef] [PubMed]
- Park, M.K.; Choi, J.K.; Kim, H.J.; Nakahata, N.; Lim, K.M.; Kim, S.Y.; Lee, C.H. Novel inhibitory effects of cardamonin on thromboxane A2-induced scratching response: Blocking of Gh/transglutaminase-2 binding to thromboxane A2 receptor. Pharmacol. Biochem. Behav. 2014, 126, 131–135. [Google Scholar] [CrossRef]
- Israf, D.A.; Khaizurin, T.A.; Syahida, A.; Lajis, N.H.; Khozirah, S. Cardamonin inhibits COX and iNOS expression via inhibition of p65NF-κB nuclear translocation and Iκ-B phosphorylation in RAW 264.7 macrophage cells. Mol. Immunol. 2007, 44, 673–679. [Google Scholar] [CrossRef]
- Chow, Y.-L.; Lee, K.-H.; Vidyadaran, S.; Lajis, N.H.; Akhtar, M.N.; Israf, D.A.; Syahida, A. Cardamonin from Alpinia rafflesiana inhibits inflammatory responses in IFN-γ/LPS-stimulated BV2 microglia via NF-κB signalling pathway. Int. Immunopharmacol. 2012, 12, 657–665. [Google Scholar] [CrossRef]
- Park, M.K.; Lee, H.J.; Choi, J.K.; Kim, H.J.; Kang, J.H.; Lee, E.J.; Kim, Y.R.; Kang, J.H.; Yoo, J.K.; Cho, H.Y.; et al. Novel anti-nociceptive effects of cardamonin via blocking expression of cyclooxygenase-2 and transglutaminase-2. Pharmacol. Biochem. Behav. 2014, 118, 10–15. [Google Scholar] [CrossRef]
- Ping, C.P.; Tengku Mohamad, T.A.S.; Akhtar, M.N.; Perimal, E.K.; Akira, A.; Israf Ali, D.A.; Sulaiman, M.R. Antinociceptive Effects of Cardamonin in Mice: Possible Involvement of TRPV₁, Glutamate, and Opioid Receptors. Molecules 2018, 23, 2237. [Google Scholar] [CrossRef]
- Wang, S.; Zhai, C.; Zhang, Y.; Yu, Y.; Zhang, Y.; Ma, L.; Li, S.; Qiao, Y. Cardamonin, a Novel Antagonist of hTRPA1 Cation Channel, Reveals Therapeutic Mechanism of Pathological Pain. Molecules 2016, 21, 1145. [Google Scholar] [CrossRef]
- Sambasevam, Y.; Omar Farouk, A.A.; Tengku Mohamad, T.A.; Sulaiman, M.R.; Bharatham, B.H.; Perimal, E.K. Cardamonin attenuates hyperalgesia and allodynia in a mouse model of chronic constriction injury-induced neuropathic pain: Possible involvement of the opioid system. Eur. J. Pharmacol. 2017, 796, 32–38. [Google Scholar] [CrossRef] [PubMed]
- Kaswan, N.K.; Mohammed Izham, N.A.B.; Tengku Mohamad, T.A.S.; Sulaiman, M.R.; Perimal, E.K. Cardamonin Modulates Neuropathic Pain through the Possible Involvement of Serotonergic 5-HT1A Receptor Pathway in CCI-Induced Neuropathic Pain Mice Model. Molecules 2021, 26, 3677. [Google Scholar] [CrossRef]
- El-Wahab, A.E.A.; Ghareeb, D.A.; Sarhan, E.E.; Abu-Serie, M.M.; El Demellawy, M.A. In vitro biological assessment of Berberis vulgaris and its active constituent, berberine: Antioxidants, anti-acetyl-cholinesterase, anti-diabetic and anticancer effects. BMC Complement. Altern. Med. 2013, 13, 218. [Google Scholar] [CrossRef]
- Hu, Q.; Peng, Z.; Li, L.; Zou, X.; Xu, L.; Gong, J.; Yi, P. The efficacy of Berberine-containing quadruple therapy on helicobacter pylori eradication in China: A systematic review and meta-analysis of randomized clinical trials. Front. Pharmacol. 2020, 10, 1694. [Google Scholar] [CrossRef] [PubMed]
- Ju, J.; Li, J.; Lin, Q.; Xu, H. Efficacy and safety of berberine for dyslipidaemias: A systematic review and meta-analysis of randomized clinical trials. Phytomedicine 2018, 50, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Chen, Q.; Wu, N.; Li, Y.; Zhang, R.; Wang, J.; Gong, D.; Zou, X.; Liu, C.; Chen, J. Berberine ameliorates spatial learning memory impairment and modulates cholinergic anti-inflammatory pathway in diabetic rats. Front. Pharmacol. 2019, 10, 1003. [Google Scholar] [CrossRef]
- Kuo, C.L.; Chi, C.W.; Liu, T.Y. The anti-inflammatory potential of berberine in vitro and in vivo. Cancer Lett. 2004, 203, 127–137. [Google Scholar] [CrossRef]
- Rezaee, R.; Monemi, A.; SadeghiBonjar, M.A.; Hashemzaei, M. Berberine alleviates paclitaxel-induced neuropathy. J. Pharmacopunct. 2019, 22, 90–94. [Google Scholar] [CrossRef]
- Hashemzaei, M.; Rezaee, R. A review on pain-relieving activity of berberine. Phytother. Res. 2021, 35, 2846–2853. [Google Scholar] [CrossRef]
- Dong, J.; Zuo, Z.; Yan, W.; Liu, W.; Zheng, Q.; Liu, X. Berberine ameliorates diabetic neuropathic pain in a rat model: Involvement of oxidative stress, inflammation, and mu-opioid receptors. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2019, 392, 1141–1149. [Google Scholar] [CrossRef]
- Yang, S.; Yu, Z.; Sun, W.; Jiang, C.; Ba, X.; Zhou, Q.; Xiong, D.; Xiao, L.; Deng, Q.; Hao, Y. The antiviral alkaloid berberine ameliorates neuropathic pain in rats with peripheral nerve injury. Acta Neurol. Belg. 2020, 120, 557–564. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Lu, M.; Pan, Q.; Fichna, J.; Zheng, L.; Wang, K.; Yu, Z.; Li, Y.; Li, K.; Song, A.; et al. Berberine improves intestinal motility and visceral pain in the mouse models mimicking diarrhea-predominant irritable bowel syndrome (IBS-D) symptoms in an opioid-receptor dependent manner. PLoS ONE 2015, 10, e0145556. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Tao, C.; Liu, Z.; Lu, M.; Pan, Q.; Zheng, L.; Li, Q.; Song, Z.; Fichna, J. A randomized clinical trial of berberine hydrochloride in patients with diarrhea-predominant irritable bowel syndrome. Phytother. Res. 2015, 29, 1822–1827. [Google Scholar] [CrossRef]
- Xu, F.; Yang, J.; Meng, B.; Zheng, J.W.; Liao, Q.; Chen, J.P.; Chen, X.W. The effect of berberine on ameliorating chronic inflammatory pain and depression. Zhonghua Yi Xue Za Zhi. 2018, 98, 1103–1108. [Google Scholar] [PubMed]
- Li, C.-L.; Tan, L.-H.; Wang, Y.-F.; Luo, C.-D.; Chen, H.-B.; Lu, Q.; Li, Y.C.; Yang, X.B.; Chen, J.N.; Liu, Y.H.; et al. Comparison of anti-inflammatory effects of berberine, and its natural oxidative and reduced derivatives from Rhizoma Coptidis in vitro and in vivo. Phytomedicine 2019, 52, 272–283. [Google Scholar] [CrossRef]
- Hussien, H.M.; Abd-Elmegied, A.; Ghareeb, D.A.; Hafez, H.S.; Ahmed, H.E.A.; El-Moneam, N.A. Neuroprotective effect of berberine against environmental heavy metals-induced neurotoxicity and Alzheimer’s-like disease in rats. Food Chem. Toxicol. 2018, 111, 432–444. [Google Scholar] [CrossRef]
- Sadraie, S.; Kiasalari, Z.; Razavian, M.; Azimi, S.; Sedighnejad, L.; Afshin-Majd, S.; Baluchnejadmojarad, T.; Roghani, M. Berberine ameliorates lipopolysaccharide-induced learning and memory deficit in the rat: Insights into underlying molecular mechanisms. Metab. Brain Dis. 2019, 34, 245–255. [Google Scholar] [CrossRef]
- Gaba, S.; Saini, A.; Singh, G.; Monga, V. An insight into the medicinal attributes of berberine derivatives: A review. Bioorg. Med. Chem. 2021, 38, 116143. [Google Scholar] [CrossRef]
- Wang, Y.X.; Liu, L.; Zeng, Q.X.; Fan, T.Y.; Jiang, J.D.; Deng, H.B.; Song, D.Q. Synthesis and Identification of Novel Berberine Derivatives as Potent Inhibitors against TNF-α-Induced NF-κB Activation. Molecules 2017, 22, 1257. [Google Scholar] [CrossRef]
- Liu, Z.; Wang, X.; Zhang, H.; Zhang, S.; Li, Y.; Liu, Y.; Peng, D. Synthesis and anti-inflammatory effects of a series of novel 9-O-substituted berberine derivatives. Med. Chem. Res. 2017, 26, 672–679. [Google Scholar] [CrossRef]
- Huang, M.Y.; Lin, J.; Huang, Z.-J.; Xu, H.-G.; Hong, J.; Sun, P.-H.; Guoa, J.-L.; Chen, W.-M. Design, synthesis and anti-inflammatory effects of novel 9-O-substituted-berberine derivatives. Med. Chem. Commun. 2016, 7, 658–666. [Google Scholar] [CrossRef]
- Ameri, A.; Simmet, T. Antagonism of the aconitine-induced inexcitability by the structurally related Aconitum alkaloids, lappaconitine and ajacine. Brain Res. 1999, 842, 332–334. [Google Scholar] [CrossRef]
- Teng, G.; Zhang, X.; Zhang, C.; Chen, L.; Sun, W.; Qiu, T.; Zhang, J. Lappaconitine trifluoroacetate contained polyvinyl alcohol nanofibrous membranes: Characterization, biological activities and transdermal application. Mater. Sci. Eng. C 2020, 108, 110515. [Google Scholar] [CrossRef]
- Sun, M.L.; Ao, J.P.; Wang, Y.R.; Huang, Q.; Li, T.F.; Li, X.Y.; Wang, Y.X. Lappaconitine, a C18-diterpenoid alkaloid, exhibits antihypersensitivity in chronic pain through stimulation of spinal dynorphin A expression. Psychopharmacology 2018, 235, 2559–2571. [Google Scholar] [CrossRef]
- Ou, S.; Zhao, Y.D.; Xiao, Z.; Wen, H.Z.; Cui, J.; Ruan, H.Z. Effect of lappaconitine on neuropathic pain mediated by P2X3 receptor in rat dorsal root ganglion. Neurochem. Int. 2011, 58, 564–573. [Google Scholar] [CrossRef] [PubMed]
- Ou, S.; Zhao, Y.D.; Xiao, Z.; Wen, H.Z.; Cui, J.; Ruan, H.Z. Study of analgesic and anti-inflammatory effects of lappaconitine gelata. J. Tradit. Chin. Med. 2009, 29, 141–145. [Google Scholar]
- Maia, J.L.; Lima-Júnior, R.C.; David, J.P.; David, J.M.; Santos, F.A.; Rao, V.S. Oleanolic Acid, a Pentacyclic Triterpene Attenuates the Mustard Oil-Induced Colonic Nociception in Mice. Biol. Pharm. Bull. 2006, 29, 82–85. [Google Scholar] [CrossRef] [PubMed]
- Park, S.-H.; Sim, Y.-B.; Kang, Y.-J.; Kim, S.-S.; Kim, C.-J.; Kim, S.-J.; Suh, H.-W. Mechanisms involved in the antinociceptive effects of orally administered oleanolic acid in the mouse. Arch. Pharm. Res. 2013, 36, 905–911. [Google Scholar] [CrossRef]
- Xuyang, L.; Guangzhi, W.; Miyang, C.; Zhan, Z. Oleanolic acid administration alleviates neuropathic pain after a peripheral nerve injury by regulating microglia polarization-mediated neuroinflammation. RSC Adv. 2020, 10, 12920–12928. [Google Scholar]
- Rali, S.; Oyedeji, O.O.; Aremu, O.O.; Oyedeji, A.O.; Nkeh-Chungag, B.N. Semisynthesis of Derivatives of Oleanolic Acid from Syzygium aromaticum and Their Antinociceptive and Anti-Inflammatory Properties. Mediat. Inflamm. 2016, 2016, 8401843. [Google Scholar] [CrossRef]
- Viana, G.; Bandeira, M.; Matos, F. Analgesic and antiinflammatory effects of chalcones isolated from Myracrodruon urundeuva allemão. Phytomedicine 2003, 10, 189–195. [Google Scholar] [CrossRef] [PubMed]
- Souza, S.M.; Aquino, L.C.; Milach, A.C., Jr.; Bandeira, M.; Nobre, M.; Viana, G. Antiinflammatory and Antiulcer Properties of Tannins from Myracrodruon urundeuva Allemão (Anacardiaceae) in Rodents. Phytother. Res. 2007, 21, 220–225. [Google Scholar] [CrossRef] [PubMed]
- Favero, F.d.F.; Grando, R.; Nonato, F.R.; Sousa, I.M.; Queiroz, N.C.; Longato, G.B.; Zafred, R.R.; Carvalho, J.E.; Spindola, H.M.; Foglio, M.A. Artemisia annua L.: Evidence of sesquiterpene lactones’ fraction antinociceptive activity. BMC Complement. Altern. Med. 2014, 14, 266. [Google Scholar]
- Burnstock, G.; Krugel, U.; Abbracchio, M.; Illes, P. Purinergic signalling: From normal behaviour to pathological brain function. Prog. Neurobiol. 2011, 95, 229–274. [Google Scholar] [CrossRef]
- Ying, M.; Liu, H.; Zhang, T.; Jiang, C.; Gong, Y.; Wu, B.; Zou, L.; Yi, Z.; Rao, S.; Li, G.; et al. Effect of artemisinin on neuropathic pain mediated by P2X4 receptor in dorsal root ganglia. Neurochem. Int. 2017, 108, 27–33. [Google Scholar] [CrossRef]
- Mahdian Dehkordi, F.; Kaboutari, J.; Zendehdel, M.; Javdani, M. The antinociceptive effect of artemisinin on the inflammatory pain and role of GABAergic and opioidergic systems. Korean J. Pain 2019, 32, 160–167. [Google Scholar] [CrossRef]
- Chandrashekar, R.; Adake, P.; Rao, S.; Santanusaha, S. Wrightia tinctoria: An overview. J. Drug Deliver. Ther. 2013, 3, 196–198. [Google Scholar]
- Jannat, T.; Hossain, M.J.; El-Shehawi, A.M.; Kuddus, M.R.; Rashid, M.A.; Albogami, S.; Jafri, I.; El-Shazly, M.; Haque, M.R. Chemical and Pharmacological Profiling of Wrightia coccinea (Roxb. Ex Hornem.) Sims Focusing Antioxidant, Cytotoxic, Antidiarrheal, Hypoglycemic, and Analgesic Properties. Molecules 2022, 27, 4024. [Google Scholar] [CrossRef]
- Nirmal, S.; Pal, S.; Mandal, S.; Patil, A. Analgesic and anti-inflammatory activity of β-sitosterol isolated from Nyctanthes arbortristis leaves. Inflammopharmacology 2012, 20, 219–224. [Google Scholar] [CrossRef]
- Saheb, S.; Desai, S.; Das, K.; Haseena, S. Hepatoprotective effect of Nigella sativa seed in sterptozotocine induced diabetic albino rats: Histological observations. Int. J. Anat. Res. 2016, 4, 2459–2463. [Google Scholar] [CrossRef]
- Abd-Elbaset, M.; Arafa, E.-S.; El Sherbiny, G.; Abdel-Bakky, M.; Elgendy, A. Thymoquinone mitigate ischemia-reperfusion-induced liver injury in rats: A pivotal role of nitric oxide signaling pathway. Arch. Pharmacol. 2017, 390, 69–76. [Google Scholar] [CrossRef] [PubMed]
- Kotb, A.; Abd-Elkareem, M.; Khalil, N.; Sayed, A.-D. Protective effect of Nigella sativa on 4-nonylphenol-induced nephrotoxicity in Clarias gariepinus. Sci. Total Environ. 2018, 619–620, 692–699. [Google Scholar] [CrossRef] [PubMed]
- Mahmud, H.; Seo, H.; Kim, S.; Islam, M.; Nam, K.; Cho, H.; Song, H. Thymoquinone (TQ) inhibits the replication of intracellular Mycobacterium tuberculosis in macrophages and modulates nitric oxide production. BMC Complement. Altern. Med. 2017, 17, 279. [Google Scholar] [CrossRef] [PubMed]
- Umar, S.; Hedaya, O.; Singh, A.; Ahmed, S. Thymoquinone inhibits TNF-α-induced inflammation and cell adhesion in rheumatoid arthritis synovial fibroblasts by ASK1 regulation. Toxicol. Appl. Pharmacol. 2015, 287, 299–305. [Google Scholar] [CrossRef]
- El-Dakhakhny, M.; Madi, N.; Lembert, N.; Ammon, H. Nigella sativa oil, nigellone and derived thymoquinone inhibit synthesis of 5-lipoxygenase products in polymorphonuclear leukocytes from rats. J. Ethnopharmacol. 2002, 81, 161–164. [Google Scholar] [CrossRef]
- Abdel-Fattah, M.; Matsumoto, K.; Watanabe, H. Antinociceptive effects of Nigella sativa oil and its major component, thymoquinone, in mice. Eur. J. Pharmacol. 2000, 400, 89–97. [Google Scholar] [CrossRef]
- Celik, F.; Göçmez, C.; Karaman, H.; Kamaşak, K.; Kaplan, I.; Akıl, E.; Tufek, A.; Guzel, A.; Uzar, E. Therapeutic Effects of Thymoquinone in a Model of Neuropathic Pain. Curr. Ther. Res. Clin. 2014, 76, 11–16. [Google Scholar] [CrossRef]
- Amin, B.; Mehdi Heravi Taheri, M.; Hosseinzadeh, H. Effects of Intraperitoneal Thymoquinone on Chronic Neuropathic Pain in Rats. Planta Med. 2014, 80, 1269–1277. [Google Scholar] [CrossRef]
- Peana, A.; Moretti, M. Pharmacological activities and applications of Salvia sclarea and Salvia desoleana essential oils. Stud. Nat. Prod. Chem. 2002, 26, 391–425. [Google Scholar]
- Peana, A.; D’Aquila, P.; Chessa, M.; Moretti, M. (−)-Linalool produces antinociception in two experimental models of pain. Eur. J. Pharmacol. 2003, 460, 37–41. [Google Scholar] [CrossRef]
- Peana, A.T.; Rubattu, P.; Piga, G.G.; Fumagalli, S.; Boatto, G.; Pippia, P.; Montis, M.G. Involvement of adenosine A1 and A2A receptors in (−)-linalool-induced antinociception. Life Sci. 2006, 78, 2471–2474. [Google Scholar] [CrossRef] [PubMed]
- Berliocchi, L.; Russo, R.; Levato, A.; Fratto, V.; Bagetta, G.; Sakurada, S.; Mercuri, N.B.; Corasaniti, M.T.; Corasaniti, M. (−)-Linalool attenuates allodynia in neuropathic pain induced by spinal nerve ligation in c57/bl6 mice. Int. Rev. Neurobiol. 2009, 85, 221–235. [Google Scholar] [PubMed]
- Katsuyama, S.; Otowa, A.; Kamio, S.; Sato, K.; Yagi, T.; Kishikawa, Y.; Komatsu, T.; Bagetta, G.; Sakurada, T.; Nakamura, H. Effect of plantar subcutaneous administration of bergamot essential oil and linalool on formalin-induced nociceptive behavior in mice. Biomed. Res. 2015, 36, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Souto-Maior, F.N.; Fonsêca, D.V.; Salgado, P.R.; Monte, L.O.; de Sousa, D.P.; de Almeida, R.N. Antinociceptive and anticonvulsant effects of the monoterpene linalool oxide. Pharm. Biol. 2017, 55, 63–67. [Google Scholar] [CrossRef] [PubMed]
- Sakurada, T.; Kuwahata, H.; Katsuyama, S.; Komatsu, T.; Morrone, L.; Corasaniti, M.E. Intraplantar injection of bergamot essential oil into the mouse hindpaw: Effects on capsaicin-induced nociceptive behaviors. Int. Rev. Neurobiol. 2009, 85, 237–248. [Google Scholar] [PubMed]
- Sakurada, T.; Mizoguchi, H.; Kuwahata, H.; Katsuyama, S.; Komatsu, T.; Morrone, L.; Corasaniti, M.T.; Bagetta, G.; Sakurada, S. Intraplantar injection of bergamot essential oil induces peripheral antinociception mediated by opioid mechanism. Pharmacol. Biochem. Behav. 2011, 97, 436–443. [Google Scholar] [CrossRef]
- Kuwahata, H.; Komatsu, T.; Katsuyama, S.; Corasaniti, M.T.; Bagetta, G.; Sakurada, S.; Sakurada, T.; Takahama, K. Peripherally injected linalool and bergamot essential oil attenuate mechanical allodynia via inhibiting spinal ERK phosphorylation. Pharmacol. Biochem. Behav. 2013, 103, 735–741. [Google Scholar] [CrossRef]
- Sulaiman, M.; Perimal, E.; Zakaria, Z.; Mokhtar, F.; Akhtar, M.; Lajis, N.; Israf, D. Preliminary analysis of the antinociceptive activity of zerumbone. Fitoterapia 2009, 80, 230–232. [Google Scholar] [CrossRef]
- Sulaiman, M.R.; Perimal, E.K.; Akhtar, M.N.; Mohamad, A.S.; Khalid, M.H.; Tasrip, N.A.; Mokhtar, F.; Zakaria, Z.A.; Lajis, N.H.; Israf, D.A. Anti-inflammatory effect of zerumbone on acute and chronic inflammation models in mice. Fitoterapia 2010, 81, 855–858. [Google Scholar] [CrossRef]
- Zulazmi, N.A.; Gopalsamy, B.; Farouk, A.K.O.; Sulaiman, M.R.; Hemabarathy Bharatham, B.; Perimal, E.K. Antiallodynic and antihyperalgesic effects of zerumbone on a mouse model of chronic constriction injury-induced neuropathic pain. Fitoterapia 2015, 105, 215–221. [Google Scholar] [CrossRef]
- Perimal, E.K.; Akhtar, M.N.; Mohamad, A.S.; Khalid, M.H.; Ming, O.H.; Khalid, S.; Tatt, L.M.; Kamaldin, M.N.; Zakaria, Z.A.; Israf, D.A.; et al. Zerumbone-induced antinociception: Involvement of the L-arginine–nitric oxide–cGMP–PKC–K+ ATP channel pathways. Basic Clin. Pharmacol. Toxicol. 2011, 108, 155–162. [Google Scholar] [CrossRef] [PubMed]
- Gopalsamy, B.; Farouk, A.; Azam, T.; Tengku, S.; Roslan, M.M.; Sulaiman, E.; Perimal, K. Antiallodynic and antihyperalgesic activities of zerumbone via the suppression of IL-1β, IL-6, and TNF-α in a mouse model of neuropathic pain. J. Pain Res. 2017, 10, 2605–2619. [Google Scholar] [CrossRef] [PubMed]
- Gopalsamy, B.; Min Chia, J.; Farouk, A.; Sulaiman, M.; Perimal, E. Zerumbone-Induced Analgesia Modulated via Potassium Channels and Opioid Receptors in Chronic Constriction Injury-Induced Neuropathic Pain. Molecules 2020, 25, 3880. [Google Scholar] [CrossRef] [PubMed]
- Zulazmi, N.; Gopalsamy, B.; Siew Min, J.; Farouk, A.; Sulaiman, M.; Hemabarathy Bharatham, B.; Perimal, E. Zerumbone Alleviates Neuropathic Pain through the Involvement of L-Arginine-Nitric Oxide-cGMP-K+ ATP Channel Pathways in Chronic Constriction Injury in Mice Model. Molecules 2017, 22, 555. [Google Scholar] [CrossRef] [PubMed]
- Siew Min Chia, J.; Farouk, A.; Tengku Mohamad, T.A.; Sulaiman, M.; Zakaria, A.H.; Perimal, E. Zerumbone Ameliorates Neuropathic Pain Symptoms via Cannabinoid and PPAR Receptors Using In Vivo and In Silico Models. Molecules 2021, 26, 3849. [Google Scholar] [CrossRef]
- Siew Min Chia, J.; Farouk, A.; Mohamad, A.; Sulaiman, M.; Perimal, E. Zerumbone alleviates chronic constriction injury-induced allodynia and hyperalgesia through serotonin 5-HT receptors. Biomed. Pharmacoth. 2016, 83, 1303–1310. [Google Scholar] [CrossRef]
- El Gabbas, Z.; Bezza, K.; Laadraoui, J.; Ait Laaradia, M.; Kebbou, A.; Oufquir, S.; Boukhira, A.; Aboufatima, R.; Chait, A. Salvia officinalis, Rosmarinic and Caffeic Acids Attenuate Neuropathic Pain and Improve Function Recovery after Sciatic Nerve Chronic Constriction in Mice. Evid.-Based Complement. Altern. Med. 2019, 2019, 1702378. [Google Scholar] [CrossRef]
- Schröder, S.; Beckmann, K.; Franconi, G.; Meyer-Hamme, G.; Friedemann, T.; Greten, H.J.; Rostock, M.; Efferth, T. Can medical herbs stimulate regeneration or neuroprotection and treat neuropathic pain in chemotherapy-induced peripheral neuropathy? Evid.-Based Complement. Altern. Med. 2013, 2013, 423713. [Google Scholar] [CrossRef]
- Bauer, J.; Kuehnl, S.; Rollinger, J.M.; Scherer, O.; Northoff, H.; Stuppner, H.; Werz, O.; Koeberle, A. Carnosol and Carnosic Acids from Salvia officinalis Inhibit Microsomal Prostaglandin E2 Synthase-1. J. Pharmacol. Exp. Ther. 2012, 342, 169–176. [Google Scholar] [CrossRef]
- Cheng, H.; Zhang, Y.; Lu, W.; Gao, X.; Xu, C.; Bao, H. Caffeic acid phenethyl ester attenuates neuropathic pain by suppressing the p38/NF-κB signal pathway in microglia. J. Pain Res. 2018, 11, 2709–2719. [Google Scholar] [CrossRef]

















































{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Natural Source | Active Principles | Synthetic Analogues | |
|---|---|---|---|
| Salvia divinorum | (Lamiaceae) | Salvinorin A | Herkinorin Kurkinorin |
| Mitragyna speciosa | (Rubiaceae) | Mitragynine, 7-OH-Mitragynine, Mitragynine pseudoindoxyl | MGM-16 MGM-9 |
| Collybia maculate | (Tricholomataceae) | Collybolide, 9-Epicollybolide | |
| Corydalis yanhusuo Corydalis bungeana | (Papaveraceae) | Corydine, Corydaline, Corynoline, L-tetrahydropalmatine (l-THP), Protopine, Dehydrocorydaline | Dehydrocorybulbine (DHCB) |
| Himenaea cangaceira | (Fabaceae) | Germacrene D, α-Humulene | |
| Ageratum conyzoides | (Asteraceae) | 5,6,7,3′,4′,5′-hexamethoxyflavone, Nobiletin, 5′methoxynobiletin, Eupalestin | |
| Arrabidaea brachypoda | (Bignoniaceae) | Brachydin A, Brachydin B, Brachydin C | |
| Nelumbo nucifera | (Nymphaeaceae) | N-methylcoclaurine, Coclaurine, O-Methylcoclaurine Neferine, | |
| Clinacanthus nutans | (Acanthaceae) | Gallic acid, Caffeic acid, Ferulic acid, Vitexin, Apigenin | |
| Azadirachta indica | (Meliaceae) | Azadirachtin A | |
| Vitex megapotamica | (Lamiaceae) | p-Coumaric acid, Isoquercitrin, Naringenin, Caffeic acid | |
| Salvia wagneriana | (Lamiaceae) | (–)-Hardwickiic | |
| Algrizea minor | (Myrtaceae) | βPinene, αPinene, Germacrene D, Bicyclogermacrene, (E)-Caryophyllene, Limonene | |
| Buddlejia globosa | (Buddlejiaceae) | Verbascoside | |
| Stachytarpheta cayennensis | (Verbenaceae) | Ipolamiide, verbascoside | |
| Tabernaemonta divaricata | (Apocynaceae) | Conolidine | DS39201083, DS54360155, DS34942424 |
| Spinacia oleracea | (Amarantacee) | Rubiscolin-6, Rubiscolin-5 | |
| Maytenus imbricata | (Celastraceae) | Tingenone | |
| Choisya ternata | (Rutaceae) | Ternanthranin | |
| Mansoa alliacea | (Bignoniaceae) | Apigenin | |
| Amomum subulatum, Boesenbergia pandurata, Alpinia rafflesiana, Alpinia katsumadai, Alpinia henryi, Campomanesia adamantium | (Zingiberaceae) (Myrtaceae) | Cardamonin | |
| Coptis and Berberis species | (Berberidaceae) | Berberine | |
| Aconitum species | (Ranunculaceae) | Lappaconitine | |
| Lantana camara, Lisgustrum lucidum, Rosmarinus officinalis | (Verbenaceae) (Oleacee) (Lamiaceae) | Oleanolic acid | |
| Myracrodruon urundeuva | (Anacardiaceae) | Urundeuvine A, B and C | |
| Artemisia annua | (Asteracee) | Artemisin | |
| Wrightia coccinea | (Apocynaceae) | β-Sitosterol Wrightiadione | |
| Nigella sativa | (Ranunculaceae) | Thymoquinone | |
| Citrus bergamia, Lavandula, Jasminum | (Rutacee) (Labiate) (Oleaceae) | (–)-Linalool | |
| Zingiber zerumbet | (Zingiberaceae) | Zerumbone | |
| Salvia officinalis | (Labiate) | Rosmarinic acid, Caffeic acid | |
| Ki (nM) ± S.D. | EC50 ± S.D. (Emax ± S.D.) a | ||||||
|---|---|---|---|---|---|---|---|
| Compound n | MOR | DOR | KOR | MOR | DOR | KOR | REF |
| 1 Salvinorin A | >10,000 | >10,000 | 18.74 ± 3.38 | >10,000 | >10,000 | 7 (104 ± 7) | [26] |
| 2 Salvinorin B | >10,000 | >10,000 | >10,000 | >10,000 | >10,000 | >10,000 | [26] |
| 13 Herkinorin | 12 ± 1 | 1170 ± 60 | 90 ± 2 | 500 ± 140 (130 ± 4) | >10,000 | 1320 ± 150 (140 ± 2) b | [47] |
| 14 Herkamide | 3.1 ± 0.4 | 810 ± 30 | 7430 ± 880 | 360 ± 60 (134 ± 5) | N.D. | N.D. b | [47] |
| 15 Kurkinorin | N.D. | N.D. | N.D. | 1.2 ± 0.6 | 74 ± 10 | >10,000 | [48] |
| pKi ± S.D. a | ||||
|---|---|---|---|---|
| Compound n | MOR | DOR | KOR | REF |
| 16 Mitragynine | 8.14 ± 0.28 | 7.22 ± 0.21 | 5.96 ± 0.22 | [58] |
| 17 7-OH-Mitragynine | 7.87 ± 0.16 | 6.81 ± 0.19 | 6.91 ± 0.07 | [58] |
| 18 Mitragynine pseudoindoxyl | 10.06 ± 0.39 | 8.52 ± 0.22 | 7.10 ± 0.32 | [58] |
| 20 9-hydroxycorynantheidine | 7.92 ± 0.05 | 4.51 ± 0.15 | 5.53 ± 0.07 | [58] |
| Ki (μM) ± S.D. a | EC50 ± S.D. (Emax ± S.D.) b | ||||
|---|---|---|---|---|---|
| Compound n | MOR | DOR | KOR | MOR | REF |
| 26 Corydine | 2.82 ± 0.61 | N.D. | N.D. | 0.51 ± 0.11 (102 ± 6) | [80] |
| 27 Corydaline | 1.23 ± 0.29 | N.D. | N.D. | 1.50 ± 0.44 (104 ± 6) | [80] |
| Ki (μM) ± S.D. a | ||||
|---|---|---|---|---|
| Compound n | MOR | DOR | KOR | REF |
| 47 O-methylcoclaurine | 2.0 ± 0.3 | 22.6 ± 3.9 | 3.5 ± 0.3 | [123] |
| 48 N-methylcoclaurine | 2.82 ± 0.61 | 20.1 ± 3.1 | 0.9 ± 0.1 | [123] |
| 49 Coclaurine | 5.0 ± 0.7 | 21.1 ± 2.0 | 2.2 ± 0.2 | [123] |
| 50 Neferine | 1.8±0.2 | 0.7 ± 0.1 | 3.3 ± 0.4 | [123] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Turnaturi, R.; Piana, S.; Spoto, S.; Costanzo, G.; Reina, L.; Pasquinucci, L.; Parenti, C. From Plant to Chemistry: Sources of Active Opioid Antinociceptive Principles for Medicinal Chemistry and Drug Design. Molecules 2023, 28, 7089. https://doi.org/10.3390/molecules28207089
Turnaturi R, Piana S, Spoto S, Costanzo G, Reina L, Pasquinucci L, Parenti C. From Plant to Chemistry: Sources of Active Opioid Antinociceptive Principles for Medicinal Chemistry and Drug Design. Molecules. 2023; 28(20):7089. https://doi.org/10.3390/molecules28207089
Chicago/Turabian StyleTurnaturi, Rita, Silvia Piana, Salvatore Spoto, Giuliana Costanzo, Lorena Reina, Lorella Pasquinucci, and Carmela Parenti. 2023. "From Plant to Chemistry: Sources of Active Opioid Antinociceptive Principles for Medicinal Chemistry and Drug Design" Molecules 28, no. 20: 7089. https://doi.org/10.3390/molecules28207089