
Two Novel Iboga-Type and an Oxindole Glucuronide Alkaloid from Tabernaemontana peduncularis Disclose Related Biosynthetic Pathways to Tabernaemontana divaricata
 , , ,
, , ,
Abstract
:
1. Introduction
2. Results and Discussion
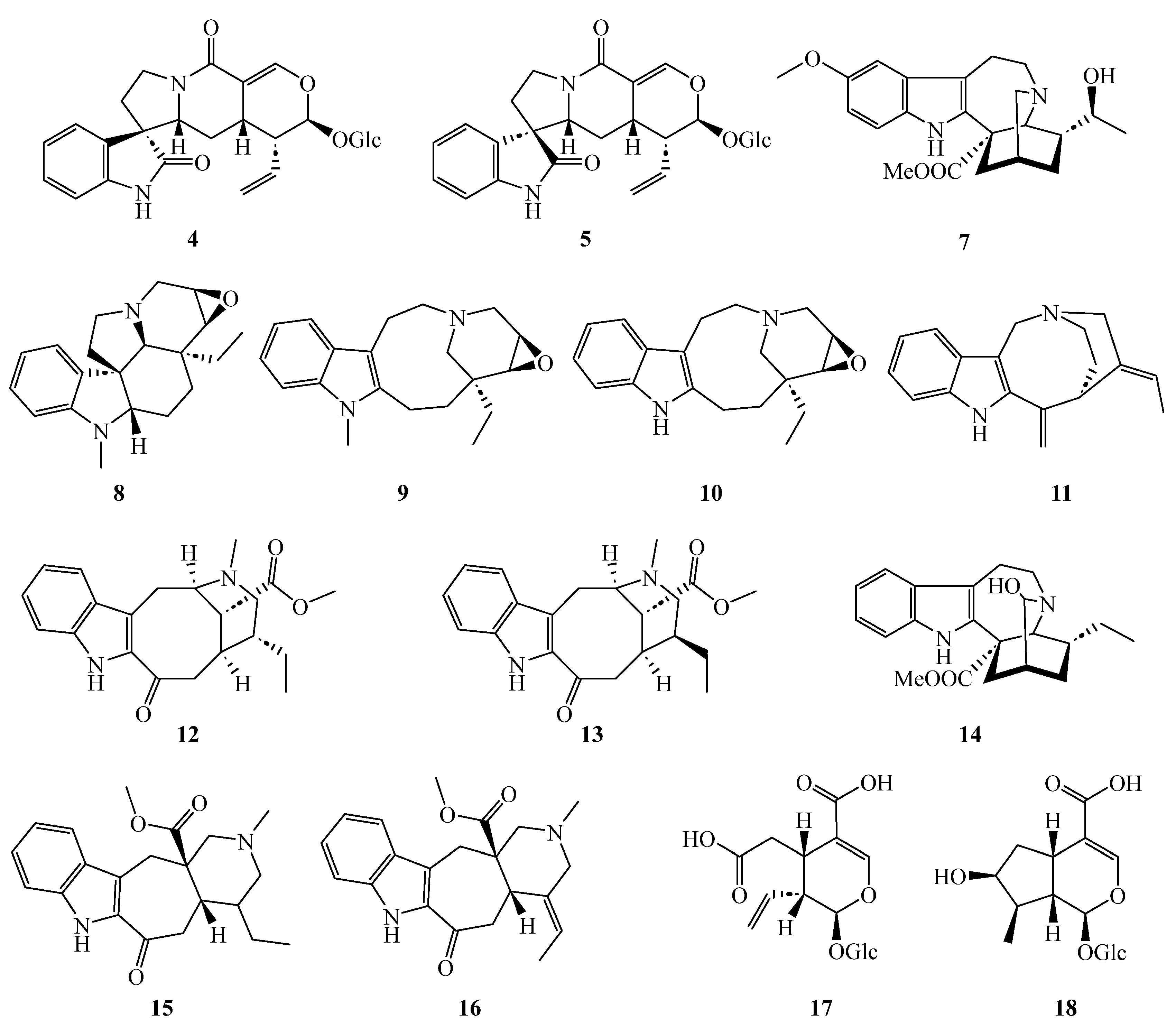
2.1. Structure Elucidation of the Novel Compounds 1, 2, and 6
2.2. Organ Specificity and Identification of the known Compounds
2.3. Biosynthetic Origins of Javaniside-Type Alkaloids 4–6
2.4. Acid Induced Rearrangement of Iboga-Type Alkaloid 1
2.5. Evaluation of Cell Viability from Crude Extracts from T. peduncularis
2.6. Cytotoxicity of Javaniside (4) and 7-epi-Javaniside (5)
2.7. Antifeedant Activities of T. peduncularis Extracts on Neonate S. littoralis Larvae
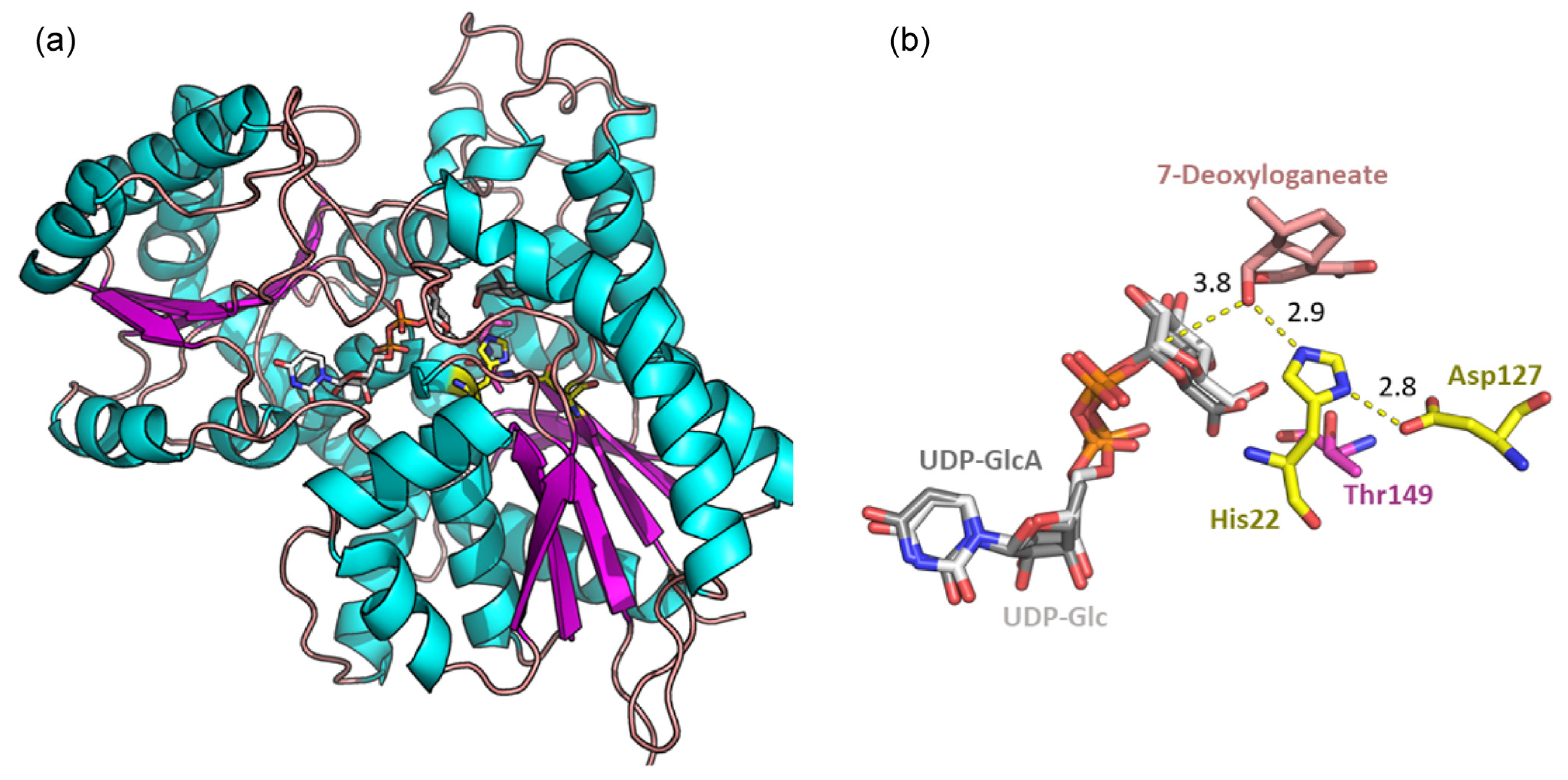
2.8. Docking Experiment
3. Experimental Section
3.1. HPLC and Chromatographic Conditions
3.2. NMR Spectroscopy
3.3. Mass Spectrometry
3.4. Plant Material
3.5. Extraction and Isolation
3.5.1. Tabernaemontana peduncularis
3.5.2. Tabernaemontana divaricata
3.6. Evaluation of Cell Viability
3.7. Cytotoxicity Assay Using SW480, CH1/PA-1, and A549 Cells
3.7.1. Cell Lines and Media
3.7.2. Assay Procedure
3.8. Antifeedant Assay
3.9. Spectroscopic Data of the Isolated Compounds
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Berger, A.; Tanuhadi, E.; Brecker, L.; Schinnerl, J.; Valant-Vetschera, K. Chemodiversity of Tryptamine-Derived Alkaloids in Six Costa Rican Palicourea Species (Rubiaceae–Palicoureeae). Phytochemistry 2017, 143, 124–131. [Google Scholar] [CrossRef]
- Dey, A.; Mukherjee, A.; Chaudhury, M. Alkaloids From Apocynaceae. In Studies in Natural Products Chemistry; Elsevier: Amsterdam, The Netherlands, 2017; Volume 52, pp. 373–488. [Google Scholar]
- Middleton, D.J. A Revision of Wrightia (Apocynaceae: Apocynoideae) in Malesia. Harv. Pap. Bot. 2005, 10, 161–182. [Google Scholar] [CrossRef]
- Zhang, Y.; Yuan, Y.-X.; Goto, M.; Guo, L.-L.; Li, X.-N.; Morris-Natschke, S.L.; Lee, K.-H.; Hao, X.-J. Taburnaemines A–I, Cytotoxic Vobasinyl-Iboga-Type Bisindole Alkaloids from Tabernaemontana corymbosa. J. Nat. Prod. 2018, 81, 562–571. [Google Scholar] [CrossRef]
- Cai, Y.-S.; Sarotti, A.M.; Zhou, T.-L.; Huang, R.; Qiu, G.; Tian, C.; Miao, Z.-H.; Mándi, A.; Kurtán, T.; Cao, S.; et al. Flabellipparicine, a Flabelliformide-Apparicine-Type Bisindole Alkaloid from Tabernaemontana divaricata. J. Nat. Prod. 2018, 81, 1976–1983. [Google Scholar] [CrossRef]
- Sim, D.S.-Y.; Teoh, W.-Y.; Sim, K.-S.; Lim, S.-H.; Thomas, N.F.; Low, Y.-Y.; Kam, T.-S. Vobatensines A–F, Cytotoxic Iboga-Vobasine Bisindoles from Tabernaemontana corymbosa. J. Nat. Prod. 2016, 79, 1048–1055. [Google Scholar] [CrossRef]
- Berger, A.; Preinfalk, A.; Robien, W.; Brecker, L.; Valant-Vetschera, K.; Schinnerl, J. New Reports on Flavonoids, Benzoic- and Chlorogenic Acids as Rare Features in the Psychotria Alliance (Rubiaceae). Biochem. Syst. Ecol. 2016, 66, 145–153. [Google Scholar] [CrossRef]
- Berger, A.; Kostyan, M.K.; Klose, S.I.; Gastegger, M.; Lorbeer, E.; Brecker, L.; Schinnerl, J. Loganin and Secologanin Derived Tryptamine–Iridoid Alkaloids from Palicourea crocea and Palicourea padifolia (Rubiaceae). Phytochemistry 2015, 116, 162–169. [Google Scholar] [CrossRef] [PubMed]
- Schinnerl, J.; Orlowska, E.A.; Lorbeer, E.; Berger, A.; Brecker, L. Alstrostines in Rubiaceae: Alstrostine A from Chassalia curviflora var. ophioxyloides and a Novel Derivative, Rudgeifoline from Rudgea cornifolia. Phytochem. Lett. 2012, 5, 586–590. [Google Scholar] [CrossRef]
- Qu, Y.; Simonescu, R.; De Luca, V. Monoterpene Indole Alkaloids from the Fruit of Tabernaemontana litoralis and Differential Alkaloid Composition in Various Fruit Components. J. Nat. Prod. 2016, 79, 3143–3147. [Google Scholar] [CrossRef] [PubMed]
- Scossa, F.; Benina, M.; Alseekh, S.; Zhang, Y.; Fernie, A. The Integration of Metabolomics and Next-Generation Sequencing Data to Elucidate the Pathways of Natural Product Metabolism in Medicinal Plants. Planta Med. 2018, 84, 855–873. [Google Scholar] [CrossRef]
- Guirimand, G.; Courdavault, V.; Lanoue, A.; Mahroug, S.; Guihur, A.; Blanc, N.; Giglioli-Guivarc’h, N.; St-Pierre, B.; Burlat, V. Strictosidine Activation in Apocynaceae: Towards a “Nuclear Time Bomb”? BMC Plant Biol. 2010, 10, 182. [Google Scholar] [CrossRef] [PubMed]
- Zèches-Hanrot, M.; Nuzillard, J.-M.; Richard, B.; Schaller, H.; Hadi, H.A.; Sévenet, T.; Men-Olivier, L. Le Alkaloids from Leaves and Stem Bark of Ervatamia peduncularis. Phytochemistry 1995, 40, 587–591. [Google Scholar] [CrossRef]
- Pretsch, E.; Bühlmann, P.; Badertscher, M. Spektroskopische Daten zur Strukturaufklärung Organischer Verbindungen; Springer: Berlin/Heidelberg, Germany, 2010; ISBN 978-3-540-76865-4. [Google Scholar]
- Farrow, S.C.; Kamileen, M.O.; Caputi, L.; Bussey, K.; Mundy, J.E.A.; McAtee, R.C.; Stephenson, C.R.J.; O’Connor, S.E. Biosynthesis of an Anti-Addiction Agent from the Iboga Plant. J. Am. Chem. Soc. 2019, 141, 12979–12983. [Google Scholar] [CrossRef]
- Karplus, M. Contact Electron-Spin Coupling of Nuclear Magnetic Moments. J. Chem. Phys. 1959, 30, 11–15. [Google Scholar] [CrossRef]
- de Nooy, A.E.J.; Besemer, A.C.; van Bekkum, H. Highly Selective Nitroxyl Radical-Mediated Oxidation of Primary Alcohol Groups in Water-Soluble Glucans. Carbohydr. Res. 1995, 269, 89–98. [Google Scholar] [CrossRef]
- Yu, Y.; Bao, M.-F.; Wu, J.; Chen, J.; Yang, Y.-R.; Schinnerl, J.; Cai, X.-H. Tabernabovines A–C: Three Monoterpenoid Indole Alkaloids from the Leaves of Tabernaemontana bovina. Org. Lett. 2019, 21, 5938–5942. [Google Scholar] [CrossRef]
- Kam, T.-S.; Sim, K.-M.; Pang, H.-S. New Bisindole Alkaloids from Tabernaemontana corymbosa. J. Nat. Prod. 2003, 66, 11–16. [Google Scholar] [CrossRef]
- Shi, B.-B.; Chen, J.; Bao, M.-F.; Zeng, Y.; Cai, X.-H. Alkaloids Isolated from Tabernaemontana bufalina Display Xanthine Oxidase Inhibitory Activity. Phytochemistry 2019, 166, 112060. [Google Scholar] [CrossRef] [PubMed]
- Tatuedom, O.K.; Kouam, S.F.; Yapna, D.B.; Ngadjui, B.T.; Green, I.R.; Choudhary, M.I.; Lantovololona, J.E.R.; Spiteller, M. Spiroalkaloids and Coumarins from the Stem Bark of Pauridiantha callicarpoides. Z. Naturforschung B 2014, 69, 747–752. [Google Scholar] [CrossRef]
- Andima, M.; Ndakala, A.; Derese, S.; Biswajyoti, S.; Hussain, A.; Yang, L.J.; Akoth, O.E.; Coghi, P.; Pal, C.; Heydenreich, M.; et al. Antileishmanial and Cytotoxic Activity of Secondary Metabolites from Taberneamontana ventricosa and Two Aloe Species. Nat. Prod. Res. 2022, 36, 1365–1369. [Google Scholar] [CrossRef]
- Han, L.; Huang, K.; Chen, C.; Zhu, W.; Ma, Y.; Hao, X.; He, H.; Zhang, Y. Taberdines L and M, Two New Alkaloids from Tabernaemontana divaricata. Nat. Prod. Res. 2022, 36, 5470–5475. [Google Scholar] [CrossRef]
- Éles, J.; Kalaus, G.; Greiner, I.; Kajtár-Peredy, M.; Szabó, P.; Keserû, G.M.; Szabó, L.; Szántay, C. Synthesis of Vinca Alkaloids and Related Compounds. 100. Stereoselective Oxidation Reactions of Compounds with the Aspidospermane and Quebrachamine Ring System. First Synthesis of Some Alkaloids Containing the Epoxy Ring. J. Org. Chem. 2002, 67, 7255–7260. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Banerjee, N.; Singamaneni, V.; Dokuparthi, S.K.; Chakrabarti, T.; Mukhopadhyay, S. Phytochemical Investigations and Evaluation of Antimutagenic Activity of the Alcoholic Extract of Glycosmis pentaphylla and Tabernaemontana coronaria by Ames Test. Nat. Prod. Res. 2018, 32, 582–587. [Google Scholar] [CrossRef] [PubMed]
- Paterna, A.; Borralho, P.M.; Gomes, S.E.; Mulhovo, S.; Rodrigues, C.M.P.; Ferreira, M.-J.U. Monoterpene Indole Alkaloid Hydrazone Derivatives with Apoptosis Inducing Activity in Human HCT116 Colon and HepG2 Liver Carcinoma Cells. Bioorg. Med. Chem. Lett. 2015, 25, 3556–3559. [Google Scholar] [CrossRef]
- Zhang, H.; Wang, X.-N.; Lin, L.-P.; Ding, J.; Yue, J.-M. Indole Alkaloids from Three Species of the Ervatamia Genus: E. officinalis, E. divaricata, and E. divaricata Gouyahua. J. Nat. Prod. 2007, 70, 54–59. [Google Scholar] [CrossRef] [PubMed]
- Zebiri, I.; Haddad, M.; Duca, L.; Sauvain, M.; Paloque, L.; Cabanillas, B.; Rengifo, E.; Behr, J.-B.; Voutquenne-Nazabadioko, L. Biological Activities of Triterpenoids from Poraqueiba sericea Stems. Nat. Prod. Res. 2017, 31, 1333–1338. [Google Scholar] [CrossRef]
- Calis, I.; Lahloub, M.F.; Sticher, O. Loganin, Loganic Acid and Periclymenoside, a New Biosidic Ester Iridoid Glucoside from Lonicera periclymenum L. (Caprifoliaceae). Helv. Chim. Acta 1984, 67, 160–165. [Google Scholar] [CrossRef]
- Ma, J.; Hecht, S.M. Javaniside, a Novel DNA Cleavage Agent from Alangium javanicum Having an Unusual Oxindole Skeleton. Chem. Commun. 2004, 4, 1190–1191. [Google Scholar] [CrossRef]
- Pham, V.C.; Ma, J.; Thomas, S.J.; Xu, Z.; Hecht, S.M. Alkaloids from Alangium javanicum and Alangium grisolleoides That Mediate Cu2+-Dependent DNA Strand Scission. J. Nat. Prod. 2005, 68, 1147–1152. [Google Scholar] [CrossRef]
- Peng, Z.-C.; He, J.; Pan, X.-G.; Zhang, J.; Wang, Y.-M.; Ye, X.-S.; Xia, C.-Y.; Lian, W.-W.; Yan, Y.; He, X.-L.; et al. Secoiridoid Dimers and Their Biogenetic Precursors from the Fruits of Cornus officinalis with Potential Therapeutic Effects on Type 2 Diabetes. Bioorg. Chem. 2021, 117, 105399. [Google Scholar] [CrossRef]
- Kornpointner, C.; Berger, A.; Traxler, F.; Hadžiabdić, A.; Massar, M.; Matek, J.; Brecker, L.; Schinnerl, J. Alkaloid and Iridoid Glucosides from Palicourea luxurians (Rubiaceae: Palicoureeae) Indicate Tryptamine- and Tryptophan-Iridoid Alkaloid Formation Apart the Strictosidine Pathway. Phytochemistry 2020, 173, 112296. [Google Scholar] [CrossRef]
- Fan, L.; Huang, X.-J.; Fan, C.-L.; Li, G.-Q.; Wu, Z.-L.; Li, S.-G.; He, Z.-D.; Wang, Y.; Ye, W.-C. Two New Oxindole Alkaloid Glycosides from the Leaves of Nauclea officinalis. Nat. Prod. Commun. 2015, 10, 2087–2090. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Tian, Z.; Zhang, Y.; Feng, X.; Li, Y.; Jiang, H. Monoterpene Indole Alkaloids in Uncaria rhynchophlla (Miq.) Jacks Chinensis and Their Chemotaxonomic Significance. Biochem. Syst. Ecol. 2020, 91, 104057. [Google Scholar] [CrossRef]
- Zheng, C.; Xia, Z.-L.; You, S.-L. Unified Mechanistic Understandings of Pictet-Spengler Reactions. Chem 2018, 4, 1952–1966. [Google Scholar] [CrossRef]
- Cong, H.-J.; Zhao, Q.; Zhang, S.-W.; Wei, J.-J.; Wang, W.-Q.; Xuan, L.-J. Terpenoid Indole Alkaloids from Mappianthus iodoides Hand.-Mazz. Phytochemistry 2014, 100, 76–85. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.-A.M.; Grzech, D.; Chung, K.; Xia, Z.; Nguyen, T.-D.; Dang, T.-T.T. Discovery of a Cytochrome P450 Enzyme Catalyzing the Formation of Spirooxindole Alkaloid Scaffold. Front. Plant Sci. 2023, 14, 1125158. [Google Scholar] [CrossRef]
- Felser, C.; Schimmer, O. Flavonoid Glycosides from Alchemilla speciosa. Planta Med. 1999, 65, 668–670. [Google Scholar] [CrossRef]
- Mbark, A.N.; Charrouf, Z.; Guillaume, D.; Kol, O. New Glycosides from Herniaria fontanesii. In Studies in Plant Science; Elsevier: Amsterdam, The Netherlands, 1999; Volume 6, pp. 314–319. [Google Scholar]
- Sun, L.-X.; Fu, W.; Ren, J.; Xu, L.; Bi, K.-S.; Wang, M.-W. Cytotoxic Constituents from Solanum lyratum. Arch. Pharm. Res. 2006, 29, 135–139. [Google Scholar] [CrossRef]
- Costa, E.M.D.; Pimenta, F.C.; Luz, W.C.; Oliveira, V.D. Selection of Filamentous Fungi of the Beauveria Genus Able to Metabolize Quercetin like Mammalian Cells. Braz. J. Microbiol. 2008, 39, 405–408. [Google Scholar] [CrossRef]
- Hodgson, E. Introduction to Biotransformation (Metabolism). In Pesticide Biotransformation and Disposition; Elsevier: Amsterdam, The Netherlands, 2012; pp. 53–72. [Google Scholar]
- Miettinen, K.; Dong, L.; Navrot, N.; Schneider, T.; Burlat, V.; Pollier, J.; Woittiez, L.; van der Krol, S.; Lugan, R.; Ilc, T.; et al. The Seco-Iridoid Pathway from Catharanthus roseus. Nat. Commun. 2014, 5, 3606. [Google Scholar] [CrossRef]
- Scudiero, D.A.; Shoemaker, R.H.; Paull, K.D.; Monks, A.; Tierney, S.; Nofziger, T.H.; Currens, M.J.; Seniff, D.; Boyd, M.R. Evaluation of a Soluble Tetrazolium/Formazan Assay for Cell Growth and Drug Sensitivity in Culture Using Human and Other Tumor Cell Lines. Cancer Res. 1988, 48, 4827–4833. [Google Scholar]
- Geran, R.S.; Greenberg, N.H.; Macdonald, M.M.; Schumacher, A.M.; Abott, B.J. Protocols for Screening Chemical Agents and Natural Products against Animal Tumors and Other Biological Systems; Cancer Chemotherapy Reports; Drug Evaluation Branch, Nat. Cancer Institute: Bethesda, MD, USA, 1972; Volume 3, pp. 1–107.
- Guo, Q.; Si, X.; Shi, Y.; Yang, H.; Liu, X.; Liang, H.; Tu, P.; Zhang, Q. Glucoconjugated Monoterpene Indole Alkaloids from Uncaria rhynchophylla. J. Nat. Prod. 2019, 82, 3288–3301. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Wu, P.; Xie, H.; Wu, G.; Wei, X. Monoterpenoid Indole Alkaloids Mediating DNA Strand Scission from Turpinia arguta. Planta Med. 2011, 77, 284–286. [Google Scholar] [CrossRef] [PubMed]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly Accurate Protein Structure Prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Eberhardt, J.; Santos-Martins, D.; Tillack, A.F.; Forli, S. AutoDock Vina 1.2.0: New Docking Methods, Expanded Force Field, and Python Bindings. J. Chem. Inf. Model 2021, 61, 3891–3898. [Google Scholar] [CrossRef] [PubMed]
- Grade Web Server. Available online: https://grade.globalphasing.org/cgi-bin/grade/server.cgi (accessed on 25 July 2023).
- Sudžuković, N.; Schinnerl, J.; Brecker, L. Phytochemical Meanings of Tetrahydro-β-Carboline Moiety in Strictosidine Derivatives. Bioorg. Med. Chem. 2016, 24, 588–595. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 1 | 2 | 6 | ||||
|---|---|---|---|---|---|---|
| pos | δC (ppm) | δH (ppm) | δC (ppm) | δH (ppm) | δC (ppm) | δH (ppm) |
| 2 | 186.0, s | 132.1, s | 181.0, s | |||
| 3 | 50.2, t | 3.62 (1H, ddd, J = 13.8, 11.3, J = 2.3) | 187.3, d | 10.53 (1H, d, J = 4.8) | 65.6, d | 4.10 (1H, J = 11.4, 3.1) |
| 3.11 (1H, ddd, J = 15.1, 4.0, J = 1.7) | ||||||
| 5 | 49.8, t | 2.81 (1H, m) | 58.1, t | 5.29 (1H, m) | 45.6, t | 4.04 (1H, m) |
| 2.64 (1H, m) | 4.23 (1H, dd, J = 12.8, 7.6) | 3.76 (1H, dd, J = 11.9, 9.9) | ||||
| 6 | 34.2, t | 2.23 (1H, m) | 20.1, t | 3.48 (2H, m) | 33.4, t | 2.42 (1H, m) |
| 2.07 (1H, ddd, J = 15.1, 2.7, J = 2.1) | 2.25 (1H, dd, J = 13.0, 7.5) | |||||
| 7 | 38.0, s | 105.9, s | 58.0, s | |||
| 8 | 141.6, s | 126.5, s | 129.5, s | |||
| 9 | 127.3, d | 7.28 (1H, d, J = 7.6) | 118.0, d | 7.44 (1H, d, J = 7.8) | 123.9, d | 7.32 (1H, d, J = 7.4) |
| 10 | 129.9 a, d | 7.35 (1H, m) | 120.0, d | 7.11 (1H, dd, J = 7.8, 7.5) | 123.7, d | 7.08 (1H, ddd, J = 7.4, 7.2, 0.7) |
| 11 | 121.7 a, d | 7.35 (1H, m) | 122.9, d | 7.17 (1H, m) | 123.9, d | 7.26 (1H, ddd, J = 7.8, 7.2, 1.0) |
| 12 | 121.3, d | 7.55 (1H, d, J = 7.8) | 112.4, d | 7.62 (1H, J = 7.8) | 111.0, d | 6.92 (1H, d, J = 7.8) |
| 13 | 150.8, s | 136.1, s | 143.5, s | |||
| 14 | 27.4, d | 1.89 (1H, m) | 33.7, d | 2.21 (1H, m) | 27.0, t | 1.39 (1H, ddd, J = 12.1, 3.9, 3.6) |
| 1.29 (1H, m) | ||||||
| 15 | 31.8, t | 1.75 (1H, m) | 29.8, t | 2.04 (1H, m) | 28.7, d | 2.99 (1H, m) |
| 1.07 (1H, m) | 1.11 (1H, m) | |||||
| 16 | 59.0, s | 55.1, s | 44.4, d | 2.63 (1H, m) | ||
| 17 | 38.4, t | 2.94 (1H, m) | 35.9, t | 2.58 (1H, d, J = 14.1) | 97.7, d | 5.52 (1H, d, J = 1.7) |
| 2.22 (1H, m) | 1.87 (1H, m) | |||||
| 18 | 11.7, q | 0.89 (3H, dd, J = 8.5, 7.2) | 11.2, q | 0.95 (3H, dd, J = 7.3, 7.2) | ||
| 19 | 27.1, t | 1.53 (1H, m) | 27.5, t | 1.42 (1H, m) | 148.3, d | 7.39 (1H, d, J = 2.3) |
| 1.42 (1H, m) | ||||||
| 20 | 38.2, d | 1.37 (1H, m) | 33.9, d | 2.03 (1H, m) | 108.9, s | |
| 21 | 56.1, d | 3.99 (1H, m) | 62.3, d | 5.07 (1H, s) | 166.0, s | |
| 22 | 172.6, s | 169.8, s | 133.8, d | 5.47 (1H, ddd, J = 17.2, 10.0, 7.2) | ||
| 23 | 120.5, t | 5.22 (1H, dd, J = 17.3, 1.6) | ||||
| 5.16 (1H, dd, J = 10.2, 1.8) | ||||||
| OMe | 52.9, q | 3.69 (3H, s) | 54.0, q | 3.85 (3H, s) | ||
| 1′ | 99.6, d | 4.66 (1H, d, J = 8.0) | ||||
| 2′ | 74.6, d | 3.18 (1H, dd, J = 8.0, 9.4 *) | ||||
| 3′ | 73.6, d | 3.41 (1H, dd, J = 9.4 *, 9.1 *) | ||||
| 4′ | 75.9, d | 3.41 (1H, dd, J = 9.1 *, 9.7 *) | ||||
| 5′ | 77.7, d | 3.58 (1H, d, J = 9.7 *) | ||||
| 6′ | 176.7, s | |||||
| Taxon | Leaves | Bark | Wood | Roots |
|---|---|---|---|---|
| T. peduncularis | 4, 5, 6 | 1, 2, 3 | / | / |
| T. divaricata | 4, 5, 7, 8, 9, 10, 11 | 11, 12, 13, 14, 15, 16 | 15, 16 | 17, 18 |
| Sample | Organ | Cell Viability ± SD | IC50 ± SD | Cytotoxic Activity |
|---|---|---|---|---|
| Crude extract | leaves | 28.47 ± 2.03 | 8.08 ± 1.56 | high |
| CHCl3 phase | stem bark | 20.79 ± 5.92 | 5.30 ± 1.02 | high |
| EtOAc phase | stem bark | 28.48 ± 1.83 | 8.12 ± 1.19 | high |
| Water phase | stem bark | 58.22 ± 10.65 | nd | nd |
| Doxorubicin | nd | 2.11 ± 0.13 | high |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Traxler, F.; Zhang, H.; Mahavorasirikul, W.; Krivanek, K.; Cai, X.-H.; Aiyakool, W.; Pfeiffer, M.; Brecker, L.; Schinnerl, J. Two Novel Iboga-Type and an Oxindole Glucuronide Alkaloid from Tabernaemontana peduncularis Disclose Related Biosynthetic Pathways to Tabernaemontana divaricata. Molecules 2023, 28, 6664. https://doi.org/10.3390/molecules28186664
Traxler F, Zhang H, Mahavorasirikul W, Krivanek K, Cai X-H, Aiyakool W, Pfeiffer M, Brecker L, Schinnerl J. Two Novel Iboga-Type and an Oxindole Glucuronide Alkaloid from Tabernaemontana peduncularis Disclose Related Biosynthetic Pathways to Tabernaemontana divaricata. Molecules. 2023; 28(18):6664. https://doi.org/10.3390/molecules28186664
Chicago/Turabian StyleTraxler, Florian, Haoqi Zhang, Wiratchanee Mahavorasirikul, Katharina Krivanek, Xiang-Hai Cai, Wichai Aiyakool, Martin Pfeiffer, Lothar Brecker, and Johann Schinnerl. 2023. "Two Novel Iboga-Type and an Oxindole Glucuronide Alkaloid from Tabernaemontana peduncularis Disclose Related Biosynthetic Pathways to Tabernaemontana divaricata" Molecules 28, no. 18: 6664. https://doi.org/10.3390/molecules28186664