
Abstract
Key message
We applied the phylogenomics to clarify the concept of rice species, aid in the identification and use of rice germplasms, and support rice biodiversity.
Abstract
Rice (genus Oryza) is one of the most important crops in the world, supporting half of the world’s population. Breeding of high-yielding and quality cultivars relies on genetic resources from both cultivated and wild species, which are collected and maintained in seed banks. Unfortunately, numerous seeds are mislabeled due to taxonomic issues or misidentifications. Here, we applied the phylogenomics of 58 complete chloroplast genomes and two hypervariable nuclear genes to determine species identity in rice seeds. Twenty-one Oryza species were identified. Conspecific relationships were determined between O. glaberrima and O. barthii, O. glumipatula and O. longistaminata, O. grandiglumis and O. alta, O. meyeriana and O. granulata, O. minuta and O. malampuzhaensis, O. nivara and O. sativa subsp. indica, and O. sativa subsp. japonica and O. rufipogon. D and L genome types were not found and the H genome type was extinct. Importantly, we evaluated the performance of four conventional plant DNA barcodes (matK, rbcL, psbA-trnH, and ITS), six rice-specific chloroplast DNA barcodes (psaJ-rpl33, trnC-rpoB, rps16-trnQ, rpl22-rps19, trnK-matK, and ndhC-trnV), two rice-specific nuclear DNA barcodes (NP78 and R22), and a chloroplast genome super DNA barcode. The latter was the most reliable marker. The six rice-specific chloroplast barcodes revealed that 17% of the 53 seed accessions from rice seed banks or field collections were mislabeled. These results are expected to clarify the concept of rice species, aid in the identification and use of rice germplasms, and support rice biodiversity.
Similar content being viewed by others

Introduction
The last 50 years witnessed an explosion in the human population, which has been supported by a three-fold global expansion in crop production (Tayyib 2013). Rice, maize, and wheat, together with some other staple crops, have been key for this expansion. The rapid increase in crop production has been achieved largely through higher yields per unit and crop intensification. Creation of higher-yielding crop varieties requires specific genes from the gene pool of the crop species and/or its close relatives, such as the semidwarfing gene in rice (sd-1) and Rht1 and Rht2 in wheat (Gale and Marshall 1973; Jennings 1964). Genetic resources are fundamental for cultivar improvement; however, most crops have suffered a loss of genetic diversity following prolonged domestication. For example, bread wheat, which originated some 8000 years ago in the Fertile Crescent, has undergone several rounds of genetic erosion (Jia et al. 2013). Genetic resources of crops and their close relatives were initially conserved ex situ in seed banks worldwide and later in situ in their homelands or nearby areas. With intense reclamation of arable land, more and more wild forms of crops and their close relatives have been lost, increasing our reliance on germplasms housed in seed banks. However, seeds in seed banks may be mislabeled due to (1) incorrect species taxonomy, (2) lack of diagnostic morphological parameters, and (3) contamination with old material. Therefore, authentication of specimens is crucial to avoid compromising research and crop production. Given that it is not easy to identify seeds based solely on morphology, DNA barcoding has come to offer a promising solution for discriminating between very similar materials.
First proposed in 2003 (Hebert et al. 2003), DNA barcoding has become a reliable technology to rapidly identify species based on short DNA fragments. In 2009, the two-locus combination of matK + rbcL was recommended as a core barcode for the identification of land plants (Hollingsworth et al. 2009). Following their first mention in 2005 (Kress et al. 2005), internal transcribed spacer of ribosomal DNA (ITS)/ITS2 and psbA-trnH were proposed as new barcodes for land plants (Chen et al. 2010; Li et al. 2011; Yan et al. 2015). A region of ycf1 was also proposed as a barcoding target owing to its high resolution (Dong et al. 2015). Due to unsatisfactory resolution of a single marker in discriminating between species, various combination schemes were assessed (Hollingsworth et al. 2009). Nowadays, the technique is successfully used to discover cryptic species (Huemer et al. 2014; Kress et al. 2009), detect illegally traded, invasive or endangered species (Lahaye et al. 2008), assess biodiversity (Sonstebo et al. 2010), and identify medicinal plants in mixtures (Howard et al. 2012). Despite these and other advancements, conventional DNA barcodes do not work in the case of extremely closely related species or only slightly diverged “species” from a recent radiation event (Hollingsworth et al. 2011). To address such instances, a DNA super barcode was proposed (Li et al. 2015). A DNA super barcode includes a complete genome or parts of a genome containing enough information to discriminate between the species of interest. The entire chloroplast or mitochondrial genomes, combinations of many genes (or regions in a genome), and assemblies of single nucleotide polymorphisms constitute examples of DNA super barcodes. With the advent of super barcodes, seeds of closely related species in seed banks can be finally assigned to the correct species or even individual haplotypes. Rice seeds require super barcodes, such as the entire chloroplast genome, to distinguish between A and C haploid genome types, which are so closely related that they cannot be resolved using common chloroplast gene fragments.
Rice belongs to the genus Oryza in the family Poaceae. The genus consists of about 26 species distributed across tropical and subtropical areas (Vaughan 1989) (Table S1). However, disputes remain regarding the relationship between O. granulata and O. meyeriana, and between O. schweinfurthiana and O. punctata. Oryza has a very short evolutionary history. It diverged from Leersia some 14 million years ago (Guo and Ge 2005) and includes eight known haploid genome types (A, B, C, E, F, G, J, K, and L) and two unknown genome types (D and H) (Aggarwal et al. 1999). The genus has been subjected to several taxonomic revisions but some issues persist (Liu et al. 2016; Lu et al. 2001; Rougerie et al. 2014; Vaughan 1989). For example, the two subspecies of the Asian rice (O. sativa), subsp. indica and subsp. japonica, are taxonomically incorrect according to International Code of Nomenclature for algae, fungi, and plants (https://www.iapt-taxon.org/nomen/main.php). Akin to African rice (O. glaberrima), its accessions are intermingled genetically with those of its wild progenitor (O. barthii, Choi et al. 2019; Li et al. 2011).
Cultivated rice is one of the most important cereal crops worldwide and it feeds more than half of the world’s population (Khush 2005). Its wild progenitors or relatives represent precious genetic resources for rice breeding and genetic improvement (Vaughan et al. 2003; Wing et al. 2005) Established genomic tools for the molecular and genetic study of O. sativa (Kim et al. 2008; Tang et al. 2010) can facilitate the correct characterization of seeds and the use of genetic resources housed in seed banks. Here, we demonstrate the effectiveness of a rice chloroplast genome super barcode for identifying rice seeds from seed banks. By employing some nuclear DNA barcodes, we also address possible faults of using the rice chloroplast genome super barcode.
Materials and methods
Seed acquisition
Fifty-three seed accessions, including two accessions of Leersia, were acquired from seed banks or collected from the field (Table 1). They proceeded mostly (41 accessions) from the International Rice Research Institute in the Philippines. Six accessions could not be traced to a particular source and three accessions were collected during our field expedition. Voucher specimens of these samples were deposited in the herbarium of the Institute of Botany, Chinese Academy of Sciences. Based on their names or field identification, the rice samples belonged to 25 species.
DNA extraction and chloroplast genome determination
Seedlings were raised from seeds in a greenhouse, harvested, and quickly dried in a convection oven at 65 °C to denature DNAase. Total genomic DNA (~ 30 mg) was extracted from dry leaves using the mCTAB method (Li et al. 2013). A library was constructed and sequenced for each sample at Beijing Novogene Bioinformatics Technology Co., Ltd, Beijing, using an Illumina HiSeq X Ten platform. Chloroplast genome reads were sorted out and the genomes were assembled de novo using SPAdes 3.9 (Bankevich et al. 2012). The generated contigs were mapped to the closest references by blastn 2.8.10 (Altschul et al. 1990), assembled with Sequencher 5.4 (Corperation)and gaps were filled by Sanger sequencing using primers reported by Dong et al. (2013).
Rice-specific DNA barcode design
Nucleotide diversity across all chloroplast genomes from all Oryza species was quantified using DnaSP (Librado and Rozas 2009). The most hypervariable regions were selected as rice-specific barcodes. Primers were designed to amplify and sequence these regions.
To determine the origins of polyploid species, two highly variable and single-copy nuclear genes were selected from 142 candidate genes (Zou et al. 2008). Fragments were amplified using specific primers. The fragments of the same sample were mixed with the chloroplast fragments and sequenced together on an Illumina HiSeq X Ten platform. Reads were extracted using known references and assembled with Sequencher 5.4.
PCR amplification and sequencing of rice-specific DNA barcodes
The PCR reaction mixture contained 1 × Taq buffer with Mg2+, 0.1 mM dNTPs, and 20 ng DNA. The PCR program included 40 cycles at 94 °C for 30 s, 55 °C for 30 s, and 72 °C for 2 min. PCR products were cleaned using PEG8000 and sequenced in both directions on an ABI 3730xl DNA Analyzer (Applied Biosystems). The sequences were assembled using Sequencher 5.4 and edited if necessary to correct some nucleotide calling mistakes.
Dataset preparation
The newly determined chloroplast genomes (Table S2) were combined with 37 chloroplast genomes (together with chloroplast fragments of three species) downloaded from GenBank (Table S3), aligned using mafft-win (Katoh and Standley 2013), and adjusted manually using Se–Al. Species delimitation, resolution comparison, and seed identification were performed with corresponding datasets using phylogenetic methods.
Dataset 1 contained 58 chloroplast genomes, representing all rice species (1–3 per species), together with three Leersia species as outgroups. Maximum parsimony analyses were carried out to identify and exclude mislabeled genomes (wrong systematic positions) or genomes of relatively low quality (longer branch lengths). This dataset was used to delimit the circumscription of species together with dataset 6 and a super barcode of Oryza.
Dataset 2 (matK), dataset 3 (rbcL), dataset 4 (psbA-trnH), and dataset 5 (ITS) represented conventional DNA barcodes. The psbA-trnH sequence is interrupted by rps19 in Poaceae. Dataset 6 represented the concatenation of two single-copy nuclear genes (N78 and R22) selected from 142 genes (Zou et al. 2008). The datasets were analyzed using phylogenetic methods to test the resolution of these candidate DNA barcodes. Dataset 7 was formed by the concatenation of six rice-specific chloroplast DNA barcodes identified in this study. This dataset was analyzed using phylogenetic methods for reliable species identification of rice seeds.
Phylogenetic analyses
Maximum parsimony
Maximum parsimony analysis was executed using PAUP version 4.0a150 (Swofford 2003). The tree search used a heuristic strategy with random stepwise addition of 100 replicates, tree bisection and reconnection branch swapping, and saving multiple trees with no more than two tree scores ≥ 5 from each replicate. Branch support for the maximum parsimony trees was assessed with 1000 bootstrap replicates. The trees were rooted using Leersia species as outgroups.
Maximum likelihood
Maximum likelihood analyses were performed using RAxML (Stamatakis 2014) with the GTR + I + G model. Branch support for the ML trees was assessed with 1000 bootstrap replicates. The trees were rooted using Leersia species as outgroups.
Bayesian inference
The best-fit substitution models were GTR + I + G and Blosum + I + G selected by running ModelFinder (Kalyaanamoorthy et al. 2017) for dataset 1 and dataset 6. Bayesian inference was assessed with MrBayes 3.2 (Fredrik et al. 2012) integrated in the PhyloSuite (Zhang et al. 2020). The Markov chain Monte Carlo process was run 2,000,000 generations and trees were sampled every 100 generations with 2 × 4 chains. Stationarity was achieved when the average standard deviation of split frequencies remained < 0.01. The first 25% of runs were discarded as burn-in. The outcomes from MrBayes were summed up by PhyloSuite and the consensus trees were rooted using Leersia species as outgroups.
Results
Rice species and their phylogenetic relationships
The phylogenetic relationships among Oryza species were reconstructed based on their complete chloroplast genomes, as well as the nuclear ITS, NP78, and R22 genes (Table S2). The eight clades in the complete chloroplast genome phylogeny matched exactly the eight genome types (Fig. 1). The species O. malampuzhaensis and O. minuta of the BC genome type formed a clade with O. punctata, indicating that a species of the B genome type was their maternal parent. O. alta, O. grandiglumis, and O. latifolia of the CD genome type and O. schweinfurthiana of the BC genome type formed a clade with species of the C genome type, suggesting their maternal parent belonged to the C genome type. Species with HJ and HK genome types did not form monophyletic clades, indicating that a species of the H genome type was their paternal parent.

The maximum likelihood strict consensus tree based on the complete chloroplast genome sequences of all species in Oryza. The figures beside branches are bootstrap values of both maximum parsimony, maximum likelihood and Bayesian analyses. Genome types are given in bold capital letters on the right side
Phylogeny based on the nuclear NP78 and R22 genes clarified the origins of allotetraploid species. Haplotypes of the same genome types formed monophyletic clades (Fig. 2). The clade comprising species with F and G genome types was located at the base, consistent with Fig. 1. The H haplotypes formed a clade independent of clades J and K, suggesting that a paternal parent with the H genome type had existed but then died out. The D haplotypes formed a clade with E haplotypes, indicating that the D genome type is a form of E. The species with a BC genome had independent origins, with O. malampuzhaensis = O. officinalis (C) × O. punctata (B) and O. schweinfurthiana = O. punctata (B) × O. eichingeri (C).

The maximum likelihood strict consensus tree based on concatenated sequences of nuclear NP78 and R22 genes of all species in Oryza. The figures beside branches are bootstrap values of both maximum parsimony, maximum likelihood and Bayesian analyses. Haplotypes are given in bold capital letters on the right side
Genetic divergence between species of the same genome type was rather small, except between O. schlechteri and O. coarctata. No significant chloroplast genome divergence was observed between O. alta and O. grandiglumis, or between O. barthii and O. glaberrima. Minor divergence was detected between O. glumipatula and O. longistaminata. In contrast, chloroplast genome divergence was clearly noted between O. sativa subsp. indica and subsp. japonica. The former formed a monophyletic clade with O. nivara, and the latter formed a monophyletic clade with O. rufipogon.
Rice-specific DNA barcodes
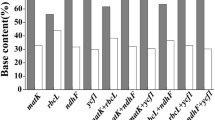
The hypervariable regions in the chloroplast genomes were identified by the sliding window method of DnaSP, and 36 regions (Table S4) were picked based on nucleotide diversity. Further evaluation of these 36 regions was carried out using the tree building method, and six high-resolution regions (psaJ-rpl33, trnC-rpoB, rps16-trnQ, rpl22-rps19, trnK-matK, and ndhC-trnV, Table 2) were finally chosen as rice-specific chloroplast DNA barcodes. While the above markers displayed higher nucleotide diversity and more variable sites than rbcL; overall, these two parameters were much higher in nuclear markers (Table 2).
Discrimination powers of conventional, rice-specific, and super DNA barcodes
The different genome types within the Oryza genus have generally diverged sufficiently for most molecular markers to discriminate between them. The resolution of the various markers is tested by the presence of more than one species per genome type. Phylogenetic methods are the most reliable way to assign a sample to a species and the following comparisons were based on the maximum parsimonious phylogenies of nearly identical samples using different molecular markers, such as matK, rbcL, psbA-trnH, ITS, NP78 + R22, rice-specific barcodes, and the super barcode. Because of narrowly or incorrectly delimited species, molecular markers cannot discriminate between the following species pairs: O. alta and O. grandiglumis (Bao and Ge 2004), O. barthii and O. glaberrima (Wang et al. 2014), O. glumipatula and O. longistaminata, O. granulata and O. meyeriana (Gong et al. 2000), O. minuta and O. malampuzhaensis, O. nivara and O. sativa subsp. indica, and O. sativa subsp. japonica and O. rufipogon.
The matK gene had an aligned length of 1417 sites with 90 parsimony-informative characters when outgroups were included. This marker failed to discriminate between species of the A, B, and C genomes (Fig. S1).
The rbcL gene had an aligned length of 1428 sites with 50 parsimony-informative characters when outgroups were considered. This marker also failed to discriminate between species of the A, B, and C genomes (Fig. S2).
The psbA-trnH region had an aligned length of 515 sites with 10 parsimony-informative characters when outgroups and partial rps19 were included. This marker could successfully identify only O. brachyantha and O. sativa subsp. indica (Fig. S3).
The nuclear ITS (including 5.8 s) had an aligned length of 713 sites with 162 parsimony-informative characters when outgroups were considered. The samples used for this marker differed slightly from those subjected to chloroplast markers because the sequences were difficult to amplify. Only one ITS copy was detected in several allotetraploid species. Phylogeny data based on ITS suggested that the H or J genome types originated from the F genome type (Fig. S4), a finding not supported by the other two nuclear genes. The ITS failed to discriminate between species of the A and C genome types.
The nuclear NP78 + R22 gene combination had an aligned length of 2218 sites with 722 parsimony-informative characters when outgroups were included. This marker combination failed to discriminate between species of the A, B, C, H, and J genome types (Fig. S5).
The rice-specific barcode consisted of six hypervariable chloroplast regions and had an aligned length of 7943 sites with 603 parsimony-informative characters when outgroups were considered. This marker combination resolved almost all species except O. punctata and O. minuta of the B genome type (Fig. 3).


The maximum parsimony strict consensus tree based on the rice-specific chloroplast DNA barcode (concatenated six hypervariable regions) sequences of all species in Oryza, demonstrating the resolution of the marker, mislabeled samples and seeds identified. Accession numbers starting with “BOP” are seeds to be identified. The figures beside branches are bootstrap values and the genome types are given in bold capital letters on the right side
Finally, the super DNA barcode of the complete chloroplast genome had an aligned length of 145,860 sites with 5048 parsimony-informative characters when outgroups were included. The super barcode exhibited the highest discriminating power, resolving all species using an insensitive but extremely reliable phylogenetic method (Fig. 1). Even though species of genome types A and C are very closely related and difficult to identify, the super barcode resolved them sufficiently well. Surprisingly, the species O. rufipogon + O. sativa subsp. japonica and O. nivara + O. sativa subsp. indica were separable using the super barcode.
Identification of seeds and mislabeled samples from seed banks
Considering that the rice-specific barcode resolved almost all rice species, we used it to identify 53 accessions of seeds from seed banks or field collections. Nine (17%) mislabeled samples were found (Fig. 3). These samples were all from species-rich genome types A and C. This was not surprising, as in the A genome type, there is still some confusion between O. rufipogon and O. nivara, and between O. glaberrima (O. barthii) and O. glumipatula. Similarly, in the C genome type, there is confusion between diploid and tetraploid O. punctata, and among tetraploid O. alta, O. latifolia, and O. minuta.
Discussion
Species delimitation and taxonomy of rice
Correct species delimitation is a prerequisite for DNA barcoding. Although considerable efforts have been made on the taxonomy of Oryza, consensus has not been reached on the number of species in the genus and some controversies remain. So far, the phylogeny of all species is incomplete. Phylogeny based on the chloroplast genome (Fig. 1) indicates that species of the E (O. australiensis) and F (O. brachyantha) genome types are monospecific and relatively isolated from other species. Species pairs have been found between O. meyeriana and O. neocaledonica of the G genome type, between O. longiglumis and O. ridleyi of the HJ genome type, and between O. coarctata of the KL genome type and O. schlechteri of the HK genome type (Lu and Ge 2003). Phylogeny based on the nuclear N78 + R22 marker (Fig. 2) revealed that the L genome did not exist, while O. coarctata belonged to the HK rather than the KL genome type. Species belonging to the HJ and HK genome types share a common paternal progenitor with the H genome, a now-extinct species originating somewhere in Irian Jaya, Indonesia or Papua New Guinea.
Major identification problems exist among species of the A, B, and C genome types. As with the H genome type, the D genome type is found only in South and Central American species, such as O. alta, O. latifolia, and O. grandiglumis, with CCDD genomes. Interestingly, the D genome type isolated from the sample BOP022669 was identified as O. latifolia and formed a clade with O. australiensis of the E genome type (Fig. 2). Phylogeny indicates that the D genome type is very likely a variant of the E genome type, if not E itself, confirming earlier results (Bao and Ge 2004; Ge et al. 1999).
There is a general correlation between molecular divergence and species delimitation (Lefébure et al. 2006). Little chloroplast genome divergence was observed between O. alta and O. grandiglumis and their conspecific nature was suggested (Bao and Ge 2004) based on nuclear genes. Considering the trivial morphological difference between O. alta and O. grandiglumis, the former becomes often a synonym of the latter instead of O. latifolia Desv., as for example on “The Plant List” (http://www.theplantlist.org/tpl1.1/record/kew-426597).
Within the BC genome type, the two Asian species O. malampuzhaensis and O. minuta originated by hybridization between O. punctata as maternal parent and O. officinalis as paternal parent (Zou et al. 2015). In contrast, for the African species O. schweinfurthiana, O. eichingeri served as maternal parent and O. punctata as paternal parent. Considering insignificant morphological differences between O. malampuzhaensis and O. minuta, the former could be regarded as a synonym of the latter. Given that O. schweinfurthiana is an allotetraploid with a different maternal parent compared to O. minuta, it should be considered a distinct species instead of merging it within O. punctata.
Misidentification of plant material is very common within the A genome type due to incorrect discrimination between species. All these species diverged within a short period by a radiation event (Wambugu et al. 2015; Zhang et al. 2014). Some species pairs exhibit neither obvious morphological difference nor remarkable genetic divergence. A first instance of confusion involves the African cultivated rice O. glaberrima and its wild progenitor O. barthii. No obvious genetic divergence has happened between their chloroplast genomes, which confirms similar results based on nuclear genes (Li et al. 2011; Wang et al. 2014). They often grow side by side in the field without ecological niche differentiation. Hence, O. barthii should be considered a synonym of O. glaberrima or a wild type.
A second confusing case involves the Asian cultivated rice O. sativa and its wild progenitors O. nivara and O. rufipogon. The Asian cultivated rice was divided into two subspecies, subsp. indica and subsp. japonica, in spite of naked names. Although the two subspecies are reproductively isolated, differ significantly in morphology and physiology, and were domesticated separately in the Himalayan mountain range and southern China (Londo et al. 2006), their taxonomic status has never been questioned. Our molecular phylogenies and almost all previous studies such as that by Wambugu et al. (2015) have confirmed that the two cultivated subspecies have the closest wild species of their own. It is very clear now that O. sativa subsp. indica is domesticated from O. nivara and that O. sativa subsp. japonica comes from O. rufipogon. Because the type of O. sativa belongs to O. sativa subsp. japonica, O. sativa must be retained in this cultivated subspecies with an autonomous name. Therefore, the two subspecies should be detached and renamed as O. sativa subsp. sativa (syn. O. sativa subsp. japonica) and O. nivara subsp. indica (syn. O. sativa subsp. indica). The names of their wild progenitors, O. nivara and O. rufipogon, have to be changed accordingly to O. sativa subsp. rufipogon (syn. O. rufipogon) and O. nivara subsp. nivara (syn. O. nivara). In 1970, a male sterile interspecific hybrid between O. nivara subsp. indica (= O. sativa subsp. indica) and O. sativa subsp. sativa (= O. sativa subsp. japonica) was discovered at a farm in Hainan province, China. The reproductive isolation between these subspecies was broken artificially and partially fertile F1 hybrid rice was used to produce fertile F2 hybrids as a new cultivar, which exhibited considerable hybrid vigor. Subsequent hybridization, however, created taxonomic problems regarding the correct identification of the two kinds of rice and their wild progenitors, resulting in many incorrectly labeled sequences being deposited in GenBank.
After synonymizing O. longistaminata under O. glumipatula and including Porteresia coarctata (Roxb.) Tateoka into Oryza (= O. coarctata Roxb.), 21 species are now recognized in the Oryza genus (supporting text S1).
Conventional DNA barcodes of rice
Three chloroplast regions (matK, psbA-trnH, and rbcL) and one nuclear region (ITS) represent conventional DNA barcodes for higher plants (Hollingsworth et al. 2009; Kress et al. 2005). Chloroplast regions perform differently in different plant groups. Here, we extracted these regions and conducted phylogenetic analyses to evaluate their suitability for species resolution. Their performance was barely satisfactory in Oryza. Generally, the matK gene offers higher resolution than rbcL, but in Oryza, it did not perform much better. Fewer than half of the 21 species were reliably (bootstrap values > 75%) resolvable. Both barcodes failed to discriminate between species of the A, B, and C genome types. Moreover, a combination of matK + rbcL did not improve the situation, because both barcodes resolved almost the same species without complementation. The psbA-trnH intergenic spacer, one of the most variable regions in chloroplast genomes, performed similarly poorly with only one identifiable species. This is probably due to the insertion of rps19, which replaced the spacer with rps19 sequences.
The nuclear ITS afforded similar resolution as conventional chloroplast regions. Although there are 10 allotetraploid species in Oryza, only one genome was detected in O. coarctata (KL), O. ridleyi (HJ), and O. schlechteri (HK). However, two kinds of sequences were observed in O. longiglumis (HJ), one of them was similar to that of O. ridleyi, and the other was similar to that of O. brachyantha, a phenomenon never reported previously. Similarly, only the C genome type was confirmed in O. alta and O. grandiglumis, whereas the B genome type defined O. malampuzhaensis. Both B and C genome types were detected in O. schweinfurthiana. The sequences deposited in GenBank include only one kind of sequence for species of the BC and CD genome types, which is probably because of concerted evolution of the ITS in relatively old tetraploids. Only newly formed tetraploids such as O. schweinfurthiana maintain both B and C genome types.
Rice-specific DNA barcodes
Most species in the Oryza genus have an evolutionary history of only a few million years. Very limited genetic variation has accumulated within such a short time and conventional DNA barcodes do not work well at species level, especially for those belonging to the A, B, and C genome types. The two most variable genes (NP78 and R22) picked out from 142 nuclear genes tested by Zou et al. (2008) served here as rice-specific nuclear DNA barcodes. Despite sequencing difficulties arising from multiple copies in tetraploid species, the combined marker performed sufficiently well. It is unlikely for the two genes to have diverged significantly in different species, thus explaining why they could discriminate between species of the same genome types.
Although species of the A, B or C genome types are very closely related, complete chloroplast genomes have accumulated enough variations to discriminate between them and all rice species are identifiable even with phylogenetic methods. Owing to the single-copy nature of chloroplast genes, mutations in chloroplast genomes become fixed and spread more quickly than those in nuclear genomes. Such mutations may not reflect a true phylogeny but are adequate for species discrimination.
The powerful performance of the complete chloroplast genome for species identification does not imply that it should be used in routine plant material identifications. There are some sensible shortcuts one can take, as a very large proportion of the chloroplast genome does not contribute much to species discrimination. The most variable regions could be an epitome of the whole genome. Here, six hypervariable regions in the chloroplast genome were selected and their combination served as rice-specific DNA barcodes. This epitome worked almost as well as the entire genome in terms of species discrimination using rice seeds from seed banks or field collections.
Identification of rice seeds
Although some seed morphological characteristics can be used successfully for seed identification, it is very difficult even for taxonomists to apply them correctly and there are species whose seeds are difficult to identify by morphology only. This explains why the wrong seeds were occasionally distributed to users. Here, we show that 17% of seeds were mislabeled, a figure high enough to deserve serious consideration. Although no algorithm has improved the assignment of specimens to species (Spouge and Mariño-Ramírez 2012), our findings suggest that phylogenetic methods offer the most reliable but also the least sensitive approach in this respect. At species level, samples in a monophyletic clade with a reasonable bootstrap support belong to the same species.
Change history
15 December 2020
In the above mentioned publication the funding section was missing. The original article has been corrected and the funding section is also published here.
References
Aggarwal RK, Brar DS, Nandi S, Huang N, Khush GS (1999) Phylogenetic relationships among Oryza species revealed by AFLP markers. Theor Appl Genet 98:1320–1328
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ (1990) Basic local alignment search tool. J Mol Biol 215:403–410
Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, Lesin VM, Nikolenko SI, Pham S, Prjibelski AD, Pyshkin AV, Sirotkin AV, Vyahhi N, Tesler G, Alekseyev MA, Pevzner PA (2012) SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol 19:455–477
Bao Y, Ge S (2004) Origin and phylogeny of Oryza species with the CD genome based on multiple-gene sequence data. Plant Syst Evol 249:55–66
Chen S, Yao H, Han J, Liu C, Song J, Shi L, Zhu Y, Ma X, Gao T, Pang X, Luo K, Li Y, Li X, Jia X, Lin Y, Leon C (2010) Validation of the ITS2 region as a novel DNA barcode for identifying medicinal plant species. PLoS ONE 5:8613
Choi JY, Zaidem M, Gutaker R, Dorph K, Singh RK, Purugganan MD (2019) The complex geography of domestication of the African rice Oryza glaberrima. PLoS Genet 15(3):e1007414
Dong W, Xu C, Cheng T, Lin K, Zhou S (2013) Sequencing angiosperm plastid genomes made easy: A complete set of universal primers and a case study on the phylogeny of saxifragales. Genome Biol Evol 5:985–997
Dong W, Xu C, Li C, Sun J, Zuo Y, Shi S, Cheng T, Guo J, Zhou S (2015) Ycf1, the most promising plastid DNA barcode of land plants. Sci Rep 5:8348
Fredrik R, Maxim T, Paul VDM, Ayres DL, Aaron D, Sebastian H, Huelsenbeck JP (2012) MrBayes 3.2: Efficient bayesian phylogenetic inference and model choice across a large model space. Syst Biol 63:539–542
Gale MD, Marshall GA (1973) Insensitivity to gibberellin in dwarf wheats. Ann Bot 37:729–735
Ge S, Sang T, Lu BR, Hong DY (1999) Phylogeny of rice genomes with emphasis on origins of allotetraploid species. PNAS 96:14400–14405
Gong Y, Borromeo T, Lu BR (2000) A biosystematic study of the Oryza meyeriana complex (Poaceae). Plant Syst Evol 224:135–151
Guo YL, Ge S (2005) Molecular phylogeny of Oryzeae (Poaceae) based on DNA sequences from chloroplast, mitochondrial, and nuclear genomes. Am J Bot 92:1548–1558
Hebert PD, Cywinska A, Ball SL, Dewaard JR (2003) Biological identifications through DNA barcodes. P Roy Soc Lond B Bio 270:313–321
Hollingsworth ML, Clark AA, Forrest LL, Richardson J, Pennington RT, Long DG, Cowan R, Chase MW, Gaudeul M, Hollingsworth PM (2009) Selecting barcoding loci for plants: evaluation of seven candidate loci with species-level sampling in three divergent groups of land plants. Mol Ecol Resour 9:439–457
Hollingsworth PM, Graham SW, Little DP (2011) Choosing and using a plant DNA barcode. PLoS ONE 6:e19254
Howard C, Socratous E, Williams S, Graham E, Fowler MR, Scott NW, Bremner PD, Slater A (2012) PlantID - DNA-based identification of multiple medicinal plants in complex mixtures. Chin Med-UK 7:18
Huemer P, Karsholt O, Mutanen M (2014) DNA barcoding as a screening tool for cryptic diversity: An example from Caryocolum, with description of a new species (Lepidoptera, Gelechiidae). ZooKeys 404:91–101
Jennings PR (1964) Plant type as a rice breeding objective 1. Crop Sci 4:13–15
Jia J, Zhao S, Kong X, Li Y, Zhao G, He W, Appels R, Pfeifer M, Tao Y, Zhang X, Jing R, Zhang C, Ma Y, Gao L, Gao C, Spannagl M, Mayer KFX, Li D, Pan S, Zheng F, Hu Q, Xia X, Li J, Liang Q, Chen J, Wicker T, Gou C, Kuang H, He G, Luo Y, Keller B, Xia Q, Lu P, Wang J, Zou H, Zhang R, Xu J, Gao J, Middleton C, Quan Z, Liu G, Wang J, Yang IWGSC, He H, Mao Z, Wang LJ (2013) Aegilops tauschii draft genome sequence reveals a gene repertoire for wheat adaptation. Nature 496:91
Kalyaanamoorthy S, Minh BQ, Wong TKF, Von Haeseler A, Jermiin LS (2017) ModelFinder: fast model selection for accurate phylogenetic estimates. Nat Methods 14:587–589
Katoh K, Standley DM (2013) MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Ecol Resour 30:772–780
Khush SG (2005) What it will take to feed 5.0 billion rice consumers in 2030. Plant Mol Biol 59:1–6
Kim H, Hurwitz B, Yu Y, Collura K, Gill N, SanMiguel P, Mullikin JC, Maher C, Nelson W, Wissotski M, Braidotti M, Kudrna D, Goicoechea JL, Stein L, Ware D, Jackson SA, Soderlund C, Wing RA (2008) Construction, alignment and analysis of twelve framework physical maps that represent the ten genome types of the genus Oryza. Genome Biol 9:R45
Kress WJ, Wurdack KJ, Zimmer EA, Weigt LA, Janzen DH (2005) Use of DNA barcodes to identify flowering plants. PNAS 102:8369–8374
Kress WJ, Erickson DL, Jones FA, Swenson NG, Perez R, Sanjur O, Bermingham E (2009) Plant DNA barcodes and a community phylogeny of a tropical forest dynamics plot in Panama. PNAS 106:18621–18626
Lahaye R, Van der Bank M, Maurin O, Duthoit S, Savolainen V (2008) A DNA barcode for the flora of the Kruger National Park (South Africa). S Afr J Bot 74:370–371
Lefébure T, Douady CJ, Gouy M, Gibert J (2006) Relationship between morphological taxonomy and molecular divergence within Crustacea: proposal of a molecular threshold to help species delimitation. Mol Phylogenet Evol 40:435–447
Li DZ, Gao LM, Li HT, Wang H, Ge XJ, Liu JQ, Chen ZD, Zhou SL, Chen SL, Yang JB, Fu cx, Zeng CX, Yan HF, Zhu YJ, Sun YS, Cehn SY, Zhao L, Wang K, Yang T, Duan GW (2011) Comparative analysis of a large dataset indicates that internal transcribed spacer (ITS) should be incorporated into the core barcode for seed plants. PNAS 108:19641–19646
Li JL, Wang S, Yu J, Wang L, Zhou SL (2013) A modified CTAB protocol for plant DNA extraction. Chin Bull Bot 48:2–78
Li X, Yang Y, Henry RJ, Rossetto M, Wang Y, Chen S (2015) Plant DNA barcoding: from gene to genome. Bio Rev Cam Philos Soc 90:157–166
Librado P, Rozas J (2009) DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25:1451–1452
Liu J, Yan HF, Ge XJ (2016) The use of DNA barcoding on recently diverged species in the genus Gentiana (Gentianaceae) in China. PLoS ONE 11:e0153008
Londo JP, Chiang YC, Hung KH, Chiang TY, Schaal BA (2006) Phylogeography of Asian wild rice, Oryza rufipogon, reveals multiple independent domestications of cultivated rice, Oryza sativa. PNAS 103:9578–9583
Lu BR, Ge S (2003) Oryza coarctata: the name that best reflects the relationships of Porteresia coarctata (Poaceae: Oryzeae). Nor J Bot 23:555–558
Lu BR, Ge S, Sang T, Chen JK, Hong DY (2001) The current taxonomy and perplexity of the genus Oryza (Poaceae). J Syst Evol 39:373–388
Rougerie R, Kitching IJ, Haxaire J, Miller SE, Hausmann A, Hebert PDN (2014) Australian Sphingidae—DNA barcodes challenge current species boundaries and distributions. PLoS ONE 9:e101108
Sonstebo JH, Gielly L, Brysting AK, Elven R, Edwards M, Haile J, Willerslev E, Coissac E, Rioux D, Sannier J, Taberlet P, Brochmann C (2010) Using next-generation sequencing for molecular reconstruction of past Arctic vegetation and climate. Mol Ecol Resour 10:1009–1018
Spouge JL, Mariño-Ramírez L (2012) The practical evaluation of DNA barcode efficacy. Methods Mol Biol 858:365–377
Stamatakis A (2014) RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30:1312–1313
Swofford DL (2003) PAUP*:phylogenetic analysis using parsimony (and other methods). Sinauer Associates, Sunderland, MA
Tang L, Zou XH, Achoundong G, Potgieter C, Second G, Zhang DY, Ge S (2010) Phylogeny and biogeography of the rice tribe (Oryzeae): evidence from combined analysis of 20 chloroplast fragments. Mol Phylogenet Evol 54:266–277
Tayyib S (2013) FAO statistical yearbook, 2012. FAO, Budhapest
Vaughan DA (1989) The genus Oryza L. current status of taxonomy. IRRI research paper series
Vaughan DA, Morishima H, Kadowaki K (2003) Diversity in the Oryza genus. Curr Opin Plant Biol 6:139–142
Wambugu PW, Brozynska M, Furtado A, Waters DL, Henry RJ (2015) Relationships of wild and domesticated rices (Oryza AA genome species) based upon whole chloroplast genome sequences. Sci Rep 5:13957
Wang M, Yu Y, Haberer G, Marri PR, Fan C, Goicoechea JL, Zuccolo A, Song X, Kudrna D, Ammiraju JS, Cossu RM, Maldonado C, Chen J, Lee S, Sisneros N, Baynast K, Golser W, Wissotski M, Kim W, Sanchez P, Ndjiondjop MN, Sanni K, Long M, Carney J, Panaud O, Wicker T, Machado CA, Chen M, Mayer KFX, Rounsley S, Wing RA (2014) The genome sequence of African rice (Oryza glaberrima) and evidence for independent domestication. Nat Genet 46:982–988
Wing RA, Ammiraju JSS, Luo M, Kim HR, Yu Y, Kudrna D, Jackson S (2005) The Oryza map alignment project: the golden path to unlocking the genetic potential of wild rice species. Plant Mol Biol 59:53–62
Yan HF, Liu YJ, Xie XF, Zhang CY, Hu CM, Hao G, Ge XJ (2015) DNA barcoding evaluation and its taxonomic implications in the species-rich genus Primula Lin China. PLoS ONE 10:e0122903
Zhang QJ, Zhu T, Xia EH, Shi C, Liu YL, Zhang Y, Liu Y, Jiang WK, Zhao YJ, Mao S, Zhang PZ, Huang H, Jiao JY, Xu PZ, Yao QY, Zeng FC, Yang LL, Gao J, Tao DY, Wang YJ, Bennetzen JL, Gao LZ (2014) Rapid diversification of five Oryza AA genomes associated with rice adaptation. PNAS 111:E4954–E4962
Zhang D, Gao F, Jakovlić I, Zou H, Zhang J, Li WX, Wang GT (2020) PhyloSuite: an integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol Ecol Resour 20:348–355
Zou XH, Zhang FM, Zhang JG, Zang LL, Tang L, Wang J, Sang T, Ge S (2008) Analysis of 142 genes resolves the rapid diversification of the rice genus. Genome Biol 9:R49
Zou XH, Du YS, Tang L, Xu XW, Doyle JJ, Sang T, Ge S (2015) Multiple origins of BBCC allopolyploid species in the rice genus (Oryza). Sci Rep 5:14876
Funding
This study was partly supported by the Strategic Priority Research Program of the Chinese Academy of Sciences, Grant No. XDA 19050303 & XDA 23080204, and the Fundamental Research Funds for the Central Public-Service Research Institute [2018JB001].
Author information
Authors and Affiliations
Contributions
Author contributions
Shiliang Zhou and Xueying Yang contributed to the study conception and design. Material preparation, data collection and analysis were performed by Wen Zhang, Yuzhe Sun, Jia Liu, Chao Xu, Xinhui Zou, Xun Chen, Yanlei Liu and Ping Wu. The first draft of the manuscript was written by Shiliang Zhou, Wen Zhang and Yuzhe Sun and all authors commented on previous versions of the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The original version of this article was revised: the funding note was missing.
Electronic supplementary material
Below is the link to the electronic supplementary material.
11103_2020_1054_MOESM1_ESM.pdf
Figure S1. The maximum parsimony strict consensus tree based on the conventional DNA barcode matK sequences of all species in Oryza, demonstrating the resolution of the marker. The figures beside branches are bootstrap values (PDF 149.8 kb)
11103_2020_1054_MOESM2_ESM.pdf
Figure S2. The maximum parsimony strict consensus tree based on the conventional DNA barcode rbcL sequences of all species in Oryza, demonstrating the resolution of the marker. The figures beside branches are bootstrap values 2 (PDF 145.6 kb)
11103_2020_1054_MOESM3_ESM.pdf
Figure S3. The maximum parsimony strict consensus tree based on the conventional DNA barcode psbA-trnH sequences of all species in Oryza, demonstrating the resolution of the marker. The figures beside branches are bootstrap values (PDF 137.2 kb)
11103_2020_1054_MOESM4_ESM.pdf
Figure S4. The maximum parsimony strict consensus tree based on the conventional DNA barcode ITS sequences of all species in Oryza, demonstrating the resolution of the marker. The figures beside branches are bootstrap values (PDF 164.5 kb)
11103_2020_1054_MOESM5_ESM.pdf
Figure S5. The maximum parsimony strict consensus tree based on the rice-specific nuclear DNA barcode of NP78 + R22 sequences of all species in Oryza, demonstrating the resolution of the marker. The figures beside branches are bootstrap values (PDF 41.4 kb)
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhang, W., Sun, Y., Liu, J. et al. DNA barcoding of Oryza: conventional, specific, and super barcodes. Plant Mol Biol 105, 215–228 (2021). https://doi.org/10.1007/s11103-020-01054-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11103-020-01054-3