WO2002060910A1 - Verfahren zur herstellung von hochreinen, tris-ortho-metallierten organo-iridium-verbindungen - Google Patents
Verfahren zur herstellung von hochreinen, tris-ortho-metallierten organo-iridium-verbindungen Download PDFInfo
- Publication number
- WO2002060910A1 WO2002060910A1 PCT/EP2002/000920 EP0200920W WO02060910A1 WO 2002060910 A1 WO2002060910 A1 WO 2002060910A1 EP 0200920 W EP0200920 W EP 0200920W WO 02060910 A1 WO02060910 A1 WO 02060910A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- iridium
- atoms
- formula
- iii
- different
- Prior art date
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F15/00—Compounds containing elements of Groups 8, 9, 10 or 18 of the Periodic System
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F15/00—Compounds containing elements of Groups 8, 9, 10 or 18 of the Periodic System
- C07F15/0006—Compounds containing elements of Groups 8, 9, 10 or 18 of the Periodic System compounds of the platinum group
- C07F15/0033—Iridium compounds
-
- H—ELECTRICITY
- H10—SEMICONDUCTOR DEVICES; ELECTRIC SOLID-STATE DEVICES NOT OTHERWISE PROVIDED FOR
- H10K—ORGANIC ELECTRIC SOLID-STATE DEVICES
- H10K50/00—Organic light-emitting devices
- H10K50/10—OLEDs or polymer light-emitting diodes [PLED]
- H10K50/11—OLEDs or polymer light-emitting diodes [PLED] characterised by the electroluminescent [EL] layers
-
- H—ELECTRICITY
- H10—SEMICONDUCTOR DEVICES; ELECTRIC SOLID-STATE DEVICES NOT OTHERWISE PROVIDED FOR
- H10K—ORGANIC ELECTRIC SOLID-STATE DEVICES
- H10K85/00—Organic materials used in the body or electrodes of devices covered by this subclass
- H10K85/30—Coordination compounds
- H10K85/341—Transition metal complexes, e.g. Ru(II)polypyridine complexes
- H10K85/342—Transition metal complexes, e.g. Ru(II)polypyridine complexes comprising iridium
Definitions
- K. Dedeian et al. describe a process starting from iridium (III) acetylacetonate and 2-phenylpyridine after the fac-tris [2- (2-pyridinyl- ⁇ N) phenyl- ⁇ C] - iridium (III) was obtained in 45% yield. Analogous to the process mentioned above, this process must also be used to remove impurities from the product by chromatographic processes, whereby - due to the solubility behavior - halogenated hydrocarbons are used [K. Dedeian, P.I. Djurovich, F.O. Garces, G. Carlson, R.J. Watts Inorg. Chem., 1991, 30, 1685-1687].
- di- ⁇ -chlorotetrakis [2- (2-pyridinyl- ⁇ N) phenyl- ⁇ C] di-iridium (III), which is initially obtained in approximately 72% yield from hydrated lridium (III) chloride and 2-phenylpyridine must be prepared [S. Spouse, KA King, PJ Spellane, RJ Watts J. Am. Chem. Soc, 1984, 06, 6647], used as starting material.
- the present invention relates to a process for the preparation of compounds I.
- R is the same or different on each occurrence of F, Cl, Br, NO 2 , CN, a straight-chain or branched or cyclic alkyl or alkoxy group with 1 to 20 C atoms, one or more non-adjacent CH 2 groups denoted by -O -, -S-, -NR 1 -, or -CONR 2 - can be replaced and where one or more H atoms can be replaced by F, or an aryl or heteroaryl group with 4 to 14 C atoms, which is replaced by a or more, non-aromatic radicals R can be substituted; where a plurality of substituents R, both on the same ring and on the two different rings together, can in turn span a further mono- or polycyclic ring system;
- R 1 and R 2 are the same or different, H or an aliphatic or aromatic hydrocarbon radical having 1 to 20 carbon atoms, a is 0, 1, 2, 3 or 4, preferably 0 or 1, b is 0, 1 or 2, preferably 0 or 1, by reacting a compound of the formula (Ia)
- radicals X, R, a and b have the meanings given under formula (I), in a dipolar protic solvent, an etherified derivative derived therefrom or N-methyl-pyrrolidinone (NMP), at temperatures in the range from 160 to 220 ° C and a concentration of the iridium-containing starting material (based on iridium) in the range from 0.05 to 1.00 mol / l and the concentration of the ligand used (aryl-pyridyl derivative) being a factor 4 to 20 higher than that of the iridium-containing starting material is for a duration of 20 to 100 hours.
- NMP N-methyl-pyrrolidinone
- Reaction media according to the invention are high-boiling dipolar protic solvents such as ethylene glycol or propylene glycol, or also higher diols or polyalcohols, such as e.g. Glycerin, or also polyether alcohols such as polyethylene glycols, for example PEG600 and PEG1000, and their etherified analogues such as e.g. Triethylene glycol dimethyl ether or poly (ethylene glycol) dimethyl ether, as well as NMP.
- high-boiling dipolar protic solvents such as ethylene glycol or propylene glycol, or also higher diols or polyalcohols, such as e.g. Glycerin, or also polyether alcohols such as polyethylene glycols, for example PEG600 and PEG1000, and their etherified analogues such as e.g. Triethylene glycol dimethyl ether or poly (ethylene glycol) dimethyl ether, as well as NMP.
- the reaction is carried out in a temperature range from 160 ° C. to 220 ° C., preferably in the range from 180 ° C. to 210 ° C.
- the concentration of the iridium-containing educt, iridium (III) acetylacetonate or a similar 1,3-diketo-chelate complex is in the range from 0.05 to 1.00 molar, preferably in the range from 0.08 to 0.25 molar.
- the molar ratio according to the invention of the iridium-containing educt, iridium (III) acetylacetonate or a similar 1,3-diketo-chelate complex to the aryl-pyridyl derivative is 1: 4 to 1:20, a ratio of 1 being preferred : 6 to 1:15, a ratio of 1: 8 to 1:12 is particularly preferred.
- the preferred concentration of the aryl-pyridyl derivative is in the range from 0.50 to 10.00 molar, particularly preferably in the range from 0.80 to 2.50 molar. In addition to lower sales, falling below the above-mentioned concentrations leads to the formation of brown by-products and thus to contamination of the product.
- the reaction is carried out within 20 to 100 h, preferably in the range from 30 to 80 h. Falling short of the above Response time has an incomplete conversion of the iridium-containing used
- the present invention further provides a process for the preparation of compounds (II)
- R " is the same or different for each F, a straight-chain or branched or cyclic alkyl group with 1 to 20 C atoms, where one or more H atoms can be replaced by F, or an aryl group with 6 to 14 C atoms, which can be substituted by one or more non-aromatic radicals R "; where a plurality of substituents R ', both on the same ring and on the two different rings together, can in turn span a further mono- or polycyclic ring system; R 1, the same or different, is an aliphatic or aromatic
- Is hydrocarbon radical having 1 to 20 carbon atoms a is 0, 1, 2, 3 or 4, preferably 0 or 1
- b is 0, 1 or 2, preferably 0 or 1, by reacting a compound of the formula (Mb)
- the ir (III) compound is iridium (III) halides or pseudohalides, such as cyanides, thiocyanates and cyanates and derivatives thereof
- the reaction is carried out at low temperatures, preferably in the range from -110 to + 10 ° C, particularly preferably in the range from -110 to -20 ° C, very particularly preferably in the range from -90 to -40 ° C.
- the implementation at -78 ° C (using an acetone / dry ice cooling bath) has proven to be favorable.
- the reaction is carried out as described below:
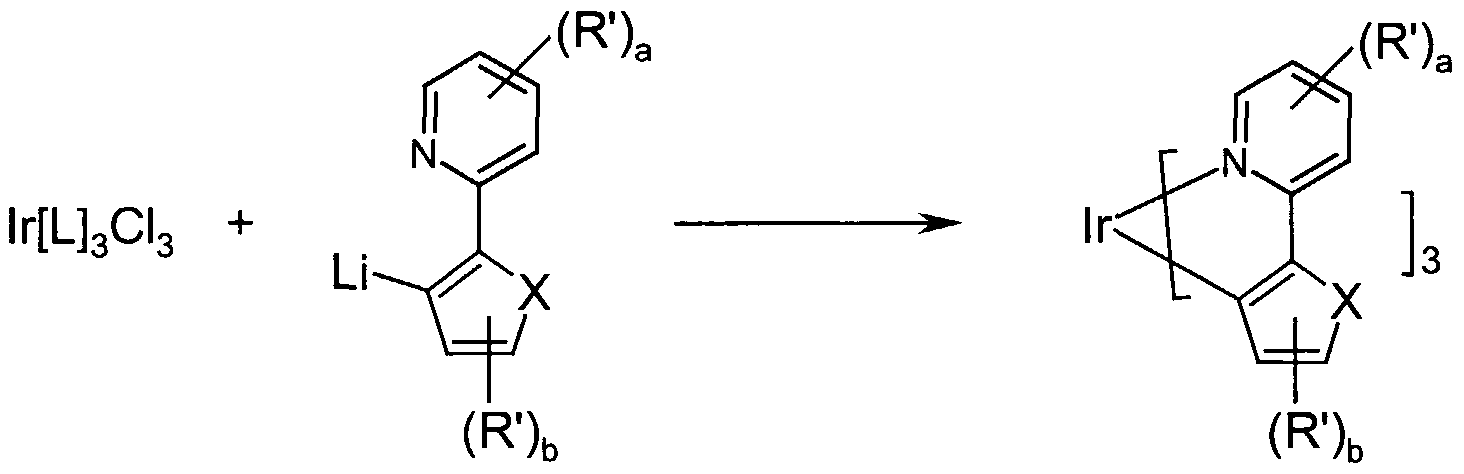
- the 2-arylpyridine or an analogous precursor according to scheme 2 below is initially at low temperatures by the action of lithium organyls such as, for. B. n-, sec- or terf-butyllithium selectively deprotonated in the 2 ' position (step 1), the addition of TMEDA (N, N, N', N'-tetramethylethylene-1, 2-diamine), 2- Hydoxyethyl-dimethyl-amine or analogs, activators known to the person skilled in the art can be advantageous.
- the aryllithium species generated in this way then governs in a second step in the sense of a salt metathetic reaction with the above-mentioned iridium (III) compounds (step 2).
- Step 1
- the crude product was taken up in 2000 ml of boiling dichloromethane, the insoluble residue was filtered off and washed twice with 200 ml of dichloromethane. The filtrate was freed from brown by-products by flash chromatography on silica gel. After adding 500 ml of methanol to the filtrate, the dichloromethane was distilled off. A yellow, microcrystalline powder was obtained.
- reaction mixture which contained the product fac-tris [2- (2-pyridinyl- ⁇ N) phenyl- ⁇ C] iridium (III) in the form of a yellow, finely crystalline precipitate, was poured into 200 ml of aqueous 1N hydrochloric acid with stirring. After stirring for 5 minutes, a
- the yield - with a purity of> 99.9% according to HPLC - was 6.04 - 6.29 g corresponding to 92.2 - 96.0%.
- Triethylene glycol dimethyl ether was replaced.
- the yield - with a purity of> 99.9% according to HPLC - was 5.90 - 6.13 g corresponding to 90.1 - 93.6%.
- Example 4 fac-Tris [2- (2-pyridinyl- ⁇ N) phenyl- ⁇ C] -iridium (III)
- the reaction mixture which contained the product ac-tris [2- (2-pyridinyl- ⁇ N) phenyl- ⁇ C] -iridium (III) in the form of a yellow, finely crystalline precipitate, was stirred into 200 ml of aqueous Poured in 1N hydrochloric acid. After stirring for 5 minutes, suction was taken off through a glass suction filter (P3), the yellow, finely crystalline precipitate was washed three times with 30 ml of aqueous 1N hydrochloric acid and five times with 30 ml of water and then under high vacuum for 5 hours at 80 ° and 2 hours at 200 ° C dried.
- P3 glass suction filter
- the reaction mixture which contained the product / ac-tris [4,5-difluoro-2- (2-pyridinyl- ⁇ N) phenyl- ⁇ C] -iridium (III) in the form of a yellow, finely crystalline precipitate
- the product was filtered off with suction through a glass suction filter (P3), the yellow, finely crystalline precipitate was washed three times with 30 ml of aqueous 1N hydrochloric acid and five times with 30 ml of water and then in a high vacuum for 5 hours at 80 ° and 2 hours at 200 ° C dried.
- the yield - with a purity of> 99.9% according to HPLC - was 7.13 - 7.37 g corresponding to 93.4 - 96.6%.
- Example 6 fac-Tris [2- (2-pyridinyl- ⁇ N) phenyl- ⁇ C] -iridium (III)
- 20.6 ml (33 mmol) of n-butyllithium 1.6 molar in n-hexane were added with stirring for 10 min.
- the deep red solution was stirred for a further 1 h at -78 ° C. and then 2.99 g of anhydrous iridium (III) chloride were added.
- the reaction mixture was allowed to warm to room temperature with stirring for 12 hours.
- the THF was then on a rotary evaporator removed, the yellow, semi-solid residue was suspended in 100 ml of ethanol and poured into 200 ml of aqueous 1N hydrochloric acid with stirring. After 5 minutes
- Example 6 Carried out analogously to Example 6, the iridium (III) chloride being replaced by 5.36 g (10 mmol) of tris (pyridine) iridium (III) chloride.
- the yield - with a purity of> 99.9% according to HPLC - was 5.83 - 6.05 g corresponding to 89.0 - 92.3%.
- Example 6 Carried out analogously to Example 6, the iridium (III) chloride being replaced by 5.66 g (10 mmol) of tris (tetrahydrothiophene) iridium (III) chloride.
- the yield - with a purity of> 99.9% according to HPLC - was 5.61 - 5.70 g corresponding to 85.7 - 87.0%.
Abstract
Description









Claims








Priority Applications (6)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| DE50202715T DE50202715D1 (de) | 2001-02-01 | 2002-01-30 | Verfahren zur herstellung von hochreinen, tris-ortho-metallierten organo-iridium-verbindungen |
| US10/470,811 US7084273B2 (en) | 2001-02-01 | 2002-01-30 | Method for the production of highly pure, tris-ortho-metalated organo-iridium compounds |
| JP2002561478A JP3984167B2 (ja) | 2001-02-01 | 2002-01-30 | 高純度のトリス−オルト−金属置換有機イリジウム化合物の製造方法 |
| EP02710817A EP1366054B1 (de) | 2001-02-01 | 2002-01-30 | Verfahren zur herstellung von hochreinen, tris-ortho-metallierten organo-iridium-verbindungen |
| KR1020037010096A KR100934147B1 (ko) | 2001-02-01 | 2002-01-30 | 고순도의 트리스-오르토-금속화된 유기 이리듐 화합물의제조방법 |
| US11/483,359 US7423151B2 (en) | 2001-02-01 | 2006-07-07 | Method for the production of highly pure tris-ortho-metalated organo-iridium compounds |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| DE10104426A DE10104426A1 (de) | 2001-02-01 | 2001-02-01 | Verfahren zur Herstellung von hochreinen, tris-ortho-metallierten Organo-Iridium-Verbindungen |
| DE10104426.7 | 2001-02-01 |
Related Child Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US10470811 A-371-Of-International | 2002-01-30 | ||
| US11/483,359 Continuation US7423151B2 (en) | 2001-02-01 | 2006-07-07 | Method for the production of highly pure tris-ortho-metalated organo-iridium compounds |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2002060910A1 true WO2002060910A1 (de) | 2002-08-08 |
Family
ID=7672422
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP2002/000920 WO2002060910A1 (de) | 2001-02-01 | 2002-01-30 | Verfahren zur herstellung von hochreinen, tris-ortho-metallierten organo-iridium-verbindungen |
Country Status (7)
| Country | Link |
|---|---|
| US (2) | US7084273B2 (de) |
| EP (1) | EP1366054B1 (de) |
| JP (1) | JP3984167B2 (de) |
| KR (1) | KR100934147B1 (de) |
| CN (2) | CN100362004C (de) |
| DE (2) | DE10104426A1 (de) |
| WO (1) | WO2002060910A1 (de) |
Cited By (114)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2004026886A2 (de) * | 2002-08-24 | 2004-04-01 | Covion Organic Semiconductors Gmbh | Rhodium-und iridium-komplexe |
| WO2004085449A1 (de) * | 2003-03-27 | 2004-10-07 | Covion Organic Semiconductors Gmbh | Verfahren zur herstellung von hochreinen organo-iridium-verbindungen |
| WO2004101707A1 (en) * | 2003-05-16 | 2004-11-25 | Isis Innovation Limited | Organic phosphorescent material and organic optoelectronic device |
| US6870054B1 (en) | 2003-12-05 | 2005-03-22 | Eastman Kodak Company | Synthesis for organometallic cyclometallated transition metal complexes |
| WO2005033244A1 (de) | 2003-09-29 | 2005-04-14 | Covion Organic Semiconductors Gmbh | Metallkomplexe |
| WO2006012023A1 (en) * | 2004-06-29 | 2006-02-02 | Eastman Kodak Company | Synthesis of organometallic cyclometallated transition metal complexes |
| WO2006018202A1 (de) * | 2004-08-12 | 2006-02-23 | Merck Patent Gmbh | Verfahren zur herstellung von iridium(iii)ketoketonaten |
| WO2007019942A1 (de) | 2005-08-18 | 2007-02-22 | Merck Patent Gmbh | Metallkomplexe |
| JP2007509872A (ja) * | 2003-10-30 | 2007-04-19 | メルク パテント ゲーエムベーハー | ヘテロレプチックオルトメタル化有機金属化合物の調製方法 |
| US7345301B2 (en) | 2003-04-15 | 2008-03-18 | Merck Patent Gmbh | Mixtures of matrix materials and organic semiconductors capable of emission, use of the same and electronic components containing said mixtures |
| CN100383150C (zh) * | 2003-05-05 | 2008-04-23 | 巴斯福股份公司 | 制造三-邻-金属化有机金属络合物的方法以及这种络合物在OLEDs中的应用 |
| EP1944309A1 (de) * | 2002-10-26 | 2008-07-16 | Merck Patent GmbH | Rhodium- und Iridium-Komplexe |
| CN100465180C (zh) * | 2003-06-07 | 2009-03-04 | 默克专利有限公司 | 制造高纯度三-和二-邻位金属化有机金属化合物的方法 |
| DE102008015526A1 (de) | 2008-03-25 | 2009-10-01 | Merck Patent Gmbh | Metallkomplexe |
| DE102008027005A1 (de) | 2008-06-05 | 2009-12-10 | Merck Patent Gmbh | Organische elektronische Vorrichtung enthaltend Metallkomplexe |
| US7700148B2 (en) | 2003-09-17 | 2010-04-20 | Toppan Printing Co., Ltd. | Electroluminescent device |
| DE102009007038A1 (de) | 2009-02-02 | 2010-08-05 | Merck Patent Gmbh | Metallkomplexe |
| WO2010097433A1 (de) | 2009-02-26 | 2010-09-02 | Basf Se | Chinonverbindungen als dotierstoff in der organischen elektronik |
| US7816531B2 (en) | 2004-07-16 | 2010-10-19 | Merck Patent Gmbh | Metal complexes |
| US7825249B2 (en) | 2004-05-19 | 2010-11-02 | Merck Patent Gmbh | Metal complexes |
| EP2251396A2 (de) | 2003-07-07 | 2010-11-17 | Merck Patent GmbH | Mischungen von organischen zur Emission befähigten Halbleitern und Matrixmaterialien, deren Verwendung und Elektronikbauteile diese enthaltend |
| DE102009041414A1 (de) | 2009-09-16 | 2011-03-17 | Merck Patent Gmbh | Metallkomplexe |
| US7923521B2 (en) | 2005-12-05 | 2011-04-12 | Merck Patent Gmbh | Process for preparing ortho-metallated metal compounds |
| DE102009049587A1 (de) | 2009-10-16 | 2011-04-21 | Merck Patent Gmbh | Metallkomplexe |
| US7981522B2 (en) | 2003-12-05 | 2011-07-19 | Merck Patent Gmbh | Organic electroluminescent element |
| WO2011141120A1 (de) | 2010-05-14 | 2011-11-17 | Merck Patent Gmbh | Metallkomplexe |
| WO2011157339A1 (de) | 2010-06-15 | 2011-12-22 | Merck Patent Gmbh | Metallkomplexe |
| WO2011157779A1 (en) | 2010-06-18 | 2011-12-22 | Basf Se | Organic electronic devices comprising a layer of a pyridine compound and a 8-hydroxyquinolinolato earth alkaline metal, or alkali metal complex |
| WO2011157790A1 (en) | 2010-06-18 | 2011-12-22 | Basf Se | Organic electronic devices comprising a layer of a dibenzofurane compound and a 8-hydroxyquinolinolato earth alkaline metal, or alkali metal complex |
| WO2012007087A1 (de) | 2010-07-16 | 2012-01-19 | Merck Patent Gmbh | Metallkomplexe |
| DE102010027317A1 (de) | 2010-07-16 | 2012-01-19 | Merck Patent Gmbh | Metallkomplexe |
| DE102010027218A1 (de) | 2010-07-15 | 2012-01-19 | Merck Patent Gmbh | Organische Komplexe enthaltend Metalle |
| WO2012038029A1 (de) | 2010-09-24 | 2012-03-29 | Merck Patent Gmbh | Phosphorhaltige metallkomplexe |
| WO2012045710A1 (en) | 2010-10-07 | 2012-04-12 | Basf Se | Phenanthro[9,10-b]furans for electronic applications |
| WO2012080052A1 (en) | 2010-12-13 | 2012-06-21 | Basf Se | Bispyrimidines for electronic applications |
| DE102012004143A1 (de) | 2011-03-31 | 2012-10-04 | Merck Patent Gmbh | Verfahren zur Herstellung von Iridium-tris(ketoketonaten) |
| WO2012130709A1 (en) | 2011-03-25 | 2012-10-04 | Basf Se | 4h-imidazo[1,2-a]imidazoles for electronic applications |
| WO2012136296A1 (de) | 2011-04-04 | 2012-10-11 | Merck Patent Gmbh | Metallkomplexe |
| US8362246B2 (en) | 2010-12-13 | 2013-01-29 | Basf Se | Bispyrimidines for electronic applications |
| US8384068B2 (en) | 2007-10-02 | 2013-02-26 | Basf Se | Use of acridine derivatives as matrix materials and/or electron blockers in OLEDs |
| WO2013068376A1 (en) | 2011-11-10 | 2013-05-16 | Basf Se | 4h-imidazo[1,2-a]imidazoles for electronic applications |
| WO2013097920A1 (en) | 2011-12-27 | 2013-07-04 | Merck Patent Gmbh | Metal complexes comprising 1,2,3-triazoles |
| US8618533B2 (en) | 2008-10-07 | 2013-12-31 | Osram Opto Semiconductors Gmbh | Siloles substituted by fused ring systems and use thereof in organic electronics |
| WO2014008982A1 (de) | 2012-07-13 | 2014-01-16 | Merck Patent Gmbh | Metallkomplexe |
| WO2014009317A1 (en) | 2012-07-10 | 2014-01-16 | Basf Se | Benzimidazo[1,2-a]benzimidazole derivatives for electronic applications |
| WO2014023377A2 (de) | 2012-08-07 | 2014-02-13 | Merck Patent Gmbh | Metallkomplexe |
| WO2014044347A1 (de) | 2012-09-20 | 2014-03-27 | Merck Patent Gmbh | Metallkomplexe |
| US8697255B2 (en) | 2007-07-05 | 2014-04-15 | Basf Se | Organic light-emitting diodes comprising at least one disilyl compound selected from disilylcarbazoles, disilyldibenzofurans, disilyldibenzothiophenes, disilyldibenzopholes, disilyldibenzothiophene S-oxides and disilyldibenzothiophene S,S-dioxides |
| WO2014072320A1 (en) | 2012-11-06 | 2014-05-15 | Basf Se | Phenoxasiline based compounds for electronic application |
| WO2014147134A1 (en) | 2013-03-20 | 2014-09-25 | Basf Se | Azabenzimidazole carbene complexes as efficiency booster in oleds |
| US8859110B2 (en) | 2008-06-20 | 2014-10-14 | Basf Se | Cyclic phosphazene compounds and use thereof in organic light emitting diodes |
| WO2015000955A1 (en) | 2013-07-02 | 2015-01-08 | Basf Se | Monosubstituted diazabenzimidazole carbene metal complexes for use in organic light emitting diodes |
| WO2015039723A1 (de) * | 2013-09-17 | 2015-03-26 | Merck Patent Gmbh | Polycyclische phenyl-pyridin iridiumkomplexe und derivate davon für oled |
| WO2015063046A1 (en) | 2013-10-31 | 2015-05-07 | Basf Se | Azadibenzothiophenes for electronic applications |
| US9079872B2 (en) | 2010-10-07 | 2015-07-14 | Basf Se | Phenanthro[9, 10-B]furans for electronic applications |
| US9142792B2 (en) | 2010-06-18 | 2015-09-22 | Basf Se | Organic electronic devices comprising a layer comprising at least one metal organic compound and at least one metal oxide |
| WO2016016791A1 (en) | 2014-07-28 | 2016-02-04 | Idemitsu Kosan Co., Ltd (Ikc) | 2,9-functionalized benzimidazolo[1,2-a]benzimidazoles as hosts for organic light emitting diodes (oleds) |
| EP2982676A1 (de) | 2014-08-07 | 2016-02-10 | Idemitsu Kosan Co., Ltd. | Benzimidazo[2,1-b]benzoxazole für elektronische Anwendungen |
| EP2993215A1 (de) | 2014-09-04 | 2016-03-09 | Idemitsu Kosan Co., Ltd. | Azabenzimidazo[2,1-a]benzimidazole für elektronische Anwendungen |
| EP3015469A1 (de) | 2014-10-30 | 2016-05-04 | Idemitsu Kosan Co., Ltd. | 5-((benz)imidazol-2-yl)benzimidazo[1,2-a]benzimidazole für elektronische anwendungen |
| WO2016079667A1 (en) | 2014-11-17 | 2016-05-26 | Idemitsu Kosan Co., Ltd. | Indole derivatives for electronic applications |
| EP3034507A1 (de) | 2014-12-15 | 2016-06-22 | Idemitsu Kosan Co., Ltd | 1-funktionalisierte Dibenzofurane und Dibenzothiophene für organische Leuchtdioden (OLEDS) |
| EP3034506A1 (de) | 2014-12-15 | 2016-06-22 | Idemitsu Kosan Co., Ltd | 4-funktionalisierte Carbazolderivate für elektronische Anwendungen |
| EP3054498A1 (de) | 2015-02-06 | 2016-08-10 | Idemitsu Kosan Co., Ltd. | Bisimidazodiazozine |
| EP3053918A1 (de) | 2015-02-06 | 2016-08-10 | Idemitsu Kosan Co., Ltd | 2-Carbazol substituierte Benzimidazole für elektronische Anwendungen |
| WO2016124304A1 (de) | 2015-02-03 | 2016-08-11 | Merck Patent Gmbh | Metallkomplexe |
| EP3061759A1 (de) | 2015-02-24 | 2016-08-31 | Idemitsu Kosan Co., Ltd | Nitrilsubstituierte dibenzofurane |
| EP3070144A1 (de) | 2015-03-17 | 2016-09-21 | Idemitsu Kosan Co., Ltd. | Siebengliedrige ringverbindungen |
| EP3072943A1 (de) | 2015-03-26 | 2016-09-28 | Idemitsu Kosan Co., Ltd. | Dibenzofuran/carbazol-substituierte benzonitrile |
| EP3075737A1 (de) | 2015-03-31 | 2016-10-05 | Idemitsu Kosan Co., Ltd | Benzimidazolo[1,2-a]benzimidazol mit aryl- oder heteroarylnitrilgruppen für organische leuchtdioden |
| EP3150606A1 (de) | 2015-10-01 | 2017-04-05 | Idemitsu Kosan Co., Ltd. | Benzimidazolo[1,2-a]benzimidazole mit benzofuran- oder benzothiophen- gruppen für organische licht emittierende dioden. |
| EP3150604A1 (de) | 2015-10-01 | 2017-04-05 | Idemitsu Kosan Co., Ltd. | Benzimidazolo[1,2-a]benzimidazol mit benzimidazolo[1,2-a]benzimidazolylgruppen, carbazolylgruppen, benzofurangruppen oder benzothiophengruppen für organische leuchtdioden |
| WO2017056053A1 (en) | 2015-10-01 | 2017-04-06 | Idemitsu Kosan Co., Ltd. | Benzimidazolo[1,2-a]benzimidazole carrying benzimidazolo[1,2-a]benzimidazolyl groups, carbazolyl groups, benzofurane groups or benzothiophene groups for organic light emitting diodes |
| WO2017056055A1 (en) | 2015-10-01 | 2017-04-06 | Idemitsu Kosan Co., Ltd. | Benzimidazolo[1,2-a]benzimidazole carrying triazine groups for organic light emitting diodes |
| WO2017078182A1 (en) | 2015-11-04 | 2017-05-11 | Idemitsu Kosan Co., Ltd. | Benzimidazole fused heteroaryls |
| WO2017093958A1 (en) | 2015-12-04 | 2017-06-08 | Idemitsu Kosan Co., Ltd. | Benzimidazolo[1,2-a]benzimidazole derivatives for organic light emitting diodes |
| WO2017109727A1 (en) | 2015-12-21 | 2017-06-29 | Idemitsu Kosan Co., Ltd. | Hetero-condensed phenylquinazolines and their use in electronic devices |
| WO2017178864A1 (en) | 2016-04-12 | 2017-10-19 | Idemitsu Kosan Co., Ltd. | Seven-membered ring compounds |
| US9806270B2 (en) | 2011-03-25 | 2017-10-31 | Udc Ireland Limited | 4H-imidazo[1,2-a]imidazoles for electronic applications |
| WO2017221999A1 (en) | 2016-06-22 | 2017-12-28 | Idemitsu Kosan Co., Ltd. | Specifically substituted benzofuro- and benzothienoquinolines for organic light emitting diodes |
| WO2018001990A1 (de) | 2016-06-30 | 2018-01-04 | Merck Patent Gmbh | Verfahren zur auftrennung von enantiomerenmischungen von metallkomplexen |
| US9862739B2 (en) | 2014-03-31 | 2018-01-09 | Udc Ireland Limited | Metal complexes, comprising carbene ligands having an O-substituted non-cyclometalated aryl group and their use in organic light emitting diodes |
| WO2018011186A1 (de) | 2016-07-14 | 2018-01-18 | Merck Patent Gmbh | Metallkomplexe |
| WO2018019688A1 (de) | 2016-07-25 | 2018-02-01 | Merck Patent Gmbh | Metallkomplexe für den einsatz als emitter in organischen elektrolumineszenzvorrichtungen |
| WO2018019687A1 (de) | 2016-07-25 | 2018-02-01 | Merck Patent Gmbh | Di- und oligonukleare metallkomplexe mit tripodalen bidentaten teilliganden sowie deren verwendung in elektronischen vorrichtungen |
| WO2018041769A1 (de) | 2016-08-30 | 2018-03-08 | Merck Patent Gmbh | Bl- und trinukleare metallkomplexe aufgebaut aus zwei miteinander verknüpften tripodalen hexadentaten liganden zur verwendung in elektrolumineszenzvorrichtungen |
| WO2018054798A1 (de) | 2016-09-21 | 2018-03-29 | Merck Patent Gmbh | Binukleare metallkomplexe für den einsatz als emitter in organischen elektrolumineszenzvorrichtungen |
| WO2018069196A1 (de) | 2016-10-12 | 2018-04-19 | Merck Patent Gmbh | Binukleare metallkomplexe sowie elektronische vorrichtungen, insbesondere organische elektrolumineszenzvorrichtungen, enthaltend diese metallkomplexe |
| WO2018069197A1 (de) | 2016-10-12 | 2018-04-19 | Merck Patent Gmbh | Metallkomplexe |
| WO2018069273A1 (de) | 2016-10-13 | 2018-04-19 | Merck Patent Gmbh | Metallkomplexe |
| WO2018077769A1 (de) | 2016-10-25 | 2018-05-03 | Merck Patent Gmbh | Metallkomplexe |
| EP3318566A1 (de) | 2012-09-20 | 2018-05-09 | UDC Ireland Limited | Azadibenzofurane für elektronische anwendungen |
| WO2018177981A1 (de) | 2017-03-29 | 2018-10-04 | Merck Patent Gmbh | Aromatische verbindungen |
| WO2019020538A1 (de) | 2017-07-25 | 2019-01-31 | Merck Patent Gmbh | Metallkomplexe |
| EP3466954A1 (de) | 2017-10-04 | 2019-04-10 | Idemitsu Kosan Co., Ltd. | Mit einem heteroatom verbrückte kondensierte phenylchinazoline |
| US10333078B2 (en) | 2014-09-26 | 2019-06-25 | Udc Ireland Limited | Fluorescent organic light emitting elements having high efficiency |
| WO2019158453A1 (de) | 2018-02-13 | 2019-08-22 | Merck Patent Gmbh | Metallkomplexe |
| US10396299B2 (en) | 2003-06-02 | 2019-08-27 | Udc Ireland Limited | Organic electroluminescent devices and metal complex compounds |
| WO2019179909A1 (de) | 2018-03-19 | 2019-09-26 | Merck Patent Gmbh | Metallkomplexe |
| EP3604477A1 (de) | 2018-07-30 | 2020-02-05 | Idemitsu Kosan Co., Ltd. | Polycyclische verbindung, organische elektrolumineszenzvorrichtung und elektronische vorrichtung |
| EP3608319A1 (de) | 2018-08-07 | 2020-02-12 | Idemitsu Kosan Co., Ltd. | Kondensierte azazyklen als organische lichtemittierende vorrichtungen und materialien zur verwendung darin |
| WO2020165064A1 (de) | 2019-02-11 | 2020-08-20 | Merck Patent Gmbh | Mononukleare iridiumkomplexe mit drei ortho-metallierten bidentaten liganden und optischer orientierungsanisotropie |
| WO2020212296A1 (de) | 2019-04-15 | 2020-10-22 | Merck Patent Gmbh | Metallkomplexe |
| US10916716B2 (en) | 2009-12-14 | 2021-02-09 | Udc Ireland Limited | Metal complexes comprising diazabenzmidazolocarbene ligands and the use thereof in OLEDS |
| US10934262B2 (en) | 2006-09-14 | 2021-03-02 | Udc Ireland Limited | Heterocyclic bridged biphenyls |
| WO2021110720A1 (de) | 2019-12-04 | 2021-06-10 | Merck Patent Gmbh | Metallkomplexe |
| US11189806B2 (en) | 2009-10-28 | 2021-11-30 | Udc Ireland Limited | Heteroleptic carbene complexes and the use thereof in organic electronics |
| US11267835B2 (en) | 2017-02-14 | 2022-03-08 | Merck Patent Gmbh | Process for preparing ortho-metallated metal compounds |
| WO2022069380A1 (de) | 2020-09-29 | 2022-04-07 | Merck Patent Gmbh | Mononukleare tripodale hexadentate iridium komplexe zur verwendung in oleds |
| US11411185B2 (en) | 2007-05-18 | 2022-08-09 | Udc Ireland Limited | Organic electroluminescent device |
| US11692131B2 (en) | 2006-04-05 | 2023-07-04 | Udc Ireland Limited | Heteroleptic transition metal-carbene complexes and their use in organic light-emitting diodes |
| US11700769B2 (en) | 2011-08-22 | 2023-07-11 | Udc Ireland Limited | Organic electroluminescent element, compound, and light emitting device, display device and lighting system, using said element |
| US11832508B2 (en) | 2009-08-31 | 2023-11-28 | Udc Ireland Limited | Organic electroluminescence device |
| EP4311849A1 (de) | 2022-07-27 | 2024-01-31 | UDC Ireland Limited | Metallkomplexe |
Families Citing this family (13)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE10104426A1 (de) * | 2001-02-01 | 2002-08-08 | Covion Organic Semiconductors | Verfahren zur Herstellung von hochreinen, tris-ortho-metallierten Organo-Iridium-Verbindungen |
| JP3969132B2 (ja) * | 2002-03-12 | 2007-09-05 | コニカミノルタホールディングス株式会社 | 有機エレクトロルミネッセンス素子及びそれを用いた表示装置 |
| DE102005032332A1 (de) * | 2005-07-08 | 2007-01-11 | Merck Patent Gmbh | Metallkomplexe |
| WO2007032203A1 (ja) | 2005-09-12 | 2007-03-22 | Nippon Steel Chemical Co., Ltd. | ホモリガンドのオルトメタル化イリジウム錯体の製造方法 |
| JP4485487B2 (ja) * | 2006-05-11 | 2010-06-23 | シャープ株式会社 | 電力増幅器 |
| EP3327100B1 (de) * | 2007-03-08 | 2020-05-06 | Universal Display Corporation | Phosphoreszierende materialien |
| US9130177B2 (en) | 2011-01-13 | 2015-09-08 | Universal Display Corporation | 5-substituted 2 phenylquinoline complexes materials for light emitting diode |
| WO2009073245A1 (en) * | 2007-12-06 | 2009-06-11 | Universal Display Corporation | Light-emitting organometallic complexes |
| WO2009073246A1 (en) * | 2007-12-06 | 2009-06-11 | Universal Display Corporation | Method for the synthesis of iridium (iii) complexes with sterically demanding ligands |
| US8993754B2 (en) | 2009-08-27 | 2015-03-31 | National Institute Of Advanced Industrial Science And Technology | Iridium complex and light emitting material formed from same |
| JP6219443B2 (ja) * | 2009-08-31 | 2017-10-25 | ユー・ディー・シー アイルランド リミテッド | 金属錯体化合物の製造方法及び有機電界発光素子 |
| JP2013527980A (ja) * | 2010-04-12 | 2013-07-04 | メルク パテント ゲーエムベーハー | 改良された性能を有する組成物 |
| US10008677B2 (en) | 2011-01-13 | 2018-06-26 | Universal Display Corporation | Materials for organic light emitting diode |
Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2001041512A1 (en) * | 1999-12-01 | 2001-06-07 | The Trustees Of Princeton University | Complexes of form l2mx as phosphorescent dopants for organic leds |
| EP1138746A1 (de) * | 2000-03-31 | 2001-10-04 | Sumitomo Chemical Company, Limited | Polymerisches fluoreszentes Material, Verfahren zu ihrer Herstellung, und lumineszentes Polymergerät worin es eingesetzt wird |
| JP2001357977A (ja) * | 2000-06-12 | 2001-12-26 | Fuji Photo Film Co Ltd | 有機電界発光素子 |
| WO2002002714A2 (en) * | 2000-06-30 | 2002-01-10 | E.I. Du Pont De Nemours And Company | Electroluminescent iridium compounds with fluorinated phenylpyridines, phenylpyrimidines, and phenylquinolines and devices made with such compounds |
| EP1175128A2 (de) * | 2000-07-17 | 2002-01-23 | Fuji Photo Film Co., Ltd. | Lichtemittierendes Element und Azolverbindung |
| WO2002015645A1 (en) * | 2000-08-11 | 2002-02-21 | The Trustees Of Princeton University | Organometallic compounds and emission-shifting organic electrophosphorescence |
Family Cites Families (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4539507A (en) | 1983-03-25 | 1985-09-03 | Eastman Kodak Company | Organic electroluminescent devices having improved power conversion efficiencies |
| US5151629A (en) | 1991-08-01 | 1992-09-29 | Eastman Kodak Company | Blue emitting internal junction organic electroluminescent device (I) |
| AU5004700A (en) | 1999-05-13 | 2000-12-05 | Trustees Of Princeton University, The | Very high efficiency organic light emitting devices based on electrophosphorescence |
| US6821645B2 (en) * | 1999-12-27 | 2004-11-23 | Fuji Photo Film Co., Ltd. | Light-emitting material comprising orthometalated iridium complex, light-emitting device, high efficiency red light-emitting device, and novel iridium complex |
| JP4460743B2 (ja) | 2000-09-29 | 2010-05-12 | 富士フイルム株式会社 | イリジウム錯体またはその互変異性体の製造方法 |
| DE10104426A1 (de) * | 2001-02-01 | 2002-08-08 | Covion Organic Semiconductors | Verfahren zur Herstellung von hochreinen, tris-ortho-metallierten Organo-Iridium-Verbindungen |
| DE10109027A1 (de) * | 2001-02-24 | 2002-09-05 | Covion Organic Semiconductors | Rhodium- und Iridium-Komplexe |
-
2001
- 2001-02-01 DE DE10104426A patent/DE10104426A1/de not_active Withdrawn
-
2002
- 2002-01-30 EP EP02710817A patent/EP1366054B1/de not_active Expired - Lifetime
- 2002-01-30 WO PCT/EP2002/000920 patent/WO2002060910A1/de active IP Right Grant
- 2002-01-30 JP JP2002561478A patent/JP3984167B2/ja not_active Expired - Fee Related
- 2002-01-30 CN CNB028042123A patent/CN100362004C/zh not_active Expired - Fee Related
- 2002-01-30 US US10/470,811 patent/US7084273B2/en not_active Expired - Fee Related
- 2002-01-30 CN CNB200510127217XA patent/CN100528885C/zh not_active Expired - Fee Related
- 2002-01-30 KR KR1020037010096A patent/KR100934147B1/ko not_active IP Right Cessation
- 2002-01-30 DE DE50202715T patent/DE50202715D1/de not_active Expired - Lifetime
-
2006
- 2006-07-07 US US11/483,359 patent/US7423151B2/en not_active Expired - Fee Related
Patent Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2001041512A1 (en) * | 1999-12-01 | 2001-06-07 | The Trustees Of Princeton University | Complexes of form l2mx as phosphorescent dopants for organic leds |
| EP1138746A1 (de) * | 2000-03-31 | 2001-10-04 | Sumitomo Chemical Company, Limited | Polymerisches fluoreszentes Material, Verfahren zu ihrer Herstellung, und lumineszentes Polymergerät worin es eingesetzt wird |
| JP2001357977A (ja) * | 2000-06-12 | 2001-12-26 | Fuji Photo Film Co Ltd | 有機電界発光素子 |
| WO2002002714A2 (en) * | 2000-06-30 | 2002-01-10 | E.I. Du Pont De Nemours And Company | Electroluminescent iridium compounds with fluorinated phenylpyridines, phenylpyrimidines, and phenylquinolines and devices made with such compounds |
| EP1175128A2 (de) * | 2000-07-17 | 2002-01-23 | Fuji Photo Film Co., Ltd. | Lichtemittierendes Element und Azolverbindung |
| WO2002015645A1 (en) * | 2000-08-11 | 2002-02-21 | The Trustees Of Princeton University | Organometallic compounds and emission-shifting organic electrophosphorescence |
Non-Patent Citations (7)
Cited By (175)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2004026886A2 (de) * | 2002-08-24 | 2004-04-01 | Covion Organic Semiconductors Gmbh | Rhodium-und iridium-komplexe |
| WO2004026886A3 (de) * | 2002-08-24 | 2004-07-01 | Covion Organic Semiconductors | Rhodium-und iridium-komplexe |
| US7883785B2 (en) | 2002-08-24 | 2011-02-08 | Merck Patent Gmbh | Rhodium and iridium complexes |
| EP2289902B1 (de) * | 2002-08-24 | 2014-04-30 | Merck Patent GmbH | Rhodium- und Iridium-Komplexe |
| CN100343263C (zh) * | 2002-08-24 | 2007-10-17 | 默克专利有限公司 | 铑和铱配合物 |
| JP2005536565A (ja) * | 2002-08-24 | 2005-12-02 | コビオン オーガニック セミコンダクターズ ゲーエムベーハー | ロジウムとイリジウムとの錯体。 |
| EP1944309A1 (de) * | 2002-10-26 | 2008-07-16 | Merck Patent GmbH | Rhodium- und Iridium-Komplexe |
| US7482450B2 (en) | 2003-03-27 | 2009-01-27 | Merck Patent Gmbh | Method for producing high-purity organoiridium compounds |
| KR101086657B1 (ko) * | 2003-03-27 | 2011-11-24 | 메르크 파텐트 게엠베하 | 고순도 유기이리듐 화합물의 제조 방법 |
| JP2006521324A (ja) * | 2003-03-27 | 2006-09-21 | メルク オレド マテリアルス ゲゼルシャフト ミット ベシュレンクテル ハフツンク | 高純度の有機イリジウム化合物を生成するための方法 |
| WO2004085449A1 (de) * | 2003-03-27 | 2004-10-07 | Covion Organic Semiconductors Gmbh | Verfahren zur herstellung von hochreinen organo-iridium-verbindungen |
| JP4680889B2 (ja) * | 2003-03-27 | 2011-05-11 | メルク パテント ゲーエムベーハー | 高純度の有機イリジウム化合物を生成するための方法 |
| CN100439380C (zh) * | 2003-03-27 | 2008-12-03 | 默克专利有限公司 | 制造高纯度有机铱化合物的方法 |
| EP2281861A2 (de) | 2003-04-15 | 2011-02-09 | Merck Patent GmbH | Mischungen von organischen zur Emission befähigten Halbleitern und Matrixmaterialien, deren Verwendung und Elektronikbauteile enthaltend diese Mischungen |
| US7345301B2 (en) | 2003-04-15 | 2008-03-18 | Merck Patent Gmbh | Mixtures of matrix materials and organic semiconductors capable of emission, use of the same and electronic components containing said mixtures |
| CN100383150C (zh) * | 2003-05-05 | 2008-04-23 | 巴斯福股份公司 | 制造三-邻-金属化有机金属络合物的方法以及这种络合物在OLEDs中的应用 |
| US8216699B2 (en) | 2003-05-16 | 2012-07-10 | Isis Innovation Limited | Organic phosphorescent material and organic optoelectronic device |
| WO2004101707A1 (en) * | 2003-05-16 | 2004-11-25 | Isis Innovation Limited | Organic phosphorescent material and organic optoelectronic device |
| US7659010B2 (en) | 2003-05-16 | 2010-02-09 | Isis Innovation Limited | Organic phosphorescent material and organic optoelectronic device |
| US11393989B2 (en) | 2003-06-02 | 2022-07-19 | Udc Ireland Limited | Organic electroluminescent devices and metal complex compounds |
| US10396299B2 (en) | 2003-06-02 | 2019-08-27 | Udc Ireland Limited | Organic electroluminescent devices and metal complex compounds |
| CN100465180C (zh) * | 2003-06-07 | 2009-03-04 | 默克专利有限公司 | 制造高纯度三-和二-邻位金属化有机金属化合物的方法 |
| US7737276B2 (en) | 2003-06-07 | 2010-06-15 | Merck Patent Gmbh | Method for producing highly purified, tris-and bis-ortho-metallated organometallic compounds |
| EP2251396A2 (de) | 2003-07-07 | 2010-11-17 | Merck Patent GmbH | Mischungen von organischen zur Emission befähigten Halbleitern und Matrixmaterialien, deren Verwendung und Elektronikbauteile diese enthaltend |
| US7700148B2 (en) | 2003-09-17 | 2010-04-20 | Toppan Printing Co., Ltd. | Electroluminescent device |
| WO2005033244A1 (de) | 2003-09-29 | 2005-04-14 | Covion Organic Semiconductors Gmbh | Metallkomplexe |
| JP2007509872A (ja) * | 2003-10-30 | 2007-04-19 | メルク パテント ゲーエムベーハー | ヘテロレプチックオルトメタル化有機金属化合物の調製方法 |
| US7745627B2 (en) | 2003-10-30 | 2010-06-29 | Merck Patent Gmbh | Method for the production of heteroleptic ortho-metallated organometallic compounds |
| US7981522B2 (en) | 2003-12-05 | 2011-07-19 | Merck Patent Gmbh | Organic electroluminescent element |
| US6870054B1 (en) | 2003-12-05 | 2005-03-22 | Eastman Kodak Company | Synthesis for organometallic cyclometallated transition metal complexes |
| JP2007513159A (ja) * | 2003-12-05 | 2007-05-24 | イーストマン コダック カンパニー | 有機金属シクロメタル化遷移金属錯体の合成 |
| US7825249B2 (en) | 2004-05-19 | 2010-11-02 | Merck Patent Gmbh | Metal complexes |
| WO2006012023A1 (en) * | 2004-06-29 | 2006-02-02 | Eastman Kodak Company | Synthesis of organometallic cyclometallated transition metal complexes |
| US7816531B2 (en) | 2004-07-16 | 2010-10-19 | Merck Patent Gmbh | Metal complexes |
| WO2006018202A1 (de) * | 2004-08-12 | 2006-02-23 | Merck Patent Gmbh | Verfahren zur herstellung von iridium(iii)ketoketonaten |
| US7589224B2 (en) | 2004-08-12 | 2009-09-15 | Merck Patent Gmbh | Method for producing iridium(III) keto ketonates |
| WO2007019942A1 (de) | 2005-08-18 | 2007-02-22 | Merck Patent Gmbh | Metallkomplexe |
| US7923521B2 (en) | 2005-12-05 | 2011-04-12 | Merck Patent Gmbh | Process for preparing ortho-metallated metal compounds |
| US11692131B2 (en) | 2006-04-05 | 2023-07-04 | Udc Ireland Limited | Heteroleptic transition metal-carbene complexes and their use in organic light-emitting diodes |
| US10934262B2 (en) | 2006-09-14 | 2021-03-02 | Udc Ireland Limited | Heterocyclic bridged biphenyls |
| US11411185B2 (en) | 2007-05-18 | 2022-08-09 | Udc Ireland Limited | Organic electroluminescent device |
| US11937504B2 (en) | 2007-05-18 | 2024-03-19 | Udc Ireland Limited | Organic electroluminescent device |
| EP3345983A1 (de) | 2007-07-05 | 2018-07-11 | UDC Ireland Limited | Verbindungen enthaltend mindestens eine disilylverbindung ausgewählt aus disilylcarbazolen, disilyldibenzofuranen, disilyldibenzothiophenen, disilyldibenzophospholen, disilyldibenzothiophen-s-oxiden und disilyldibenzothiophen-s, s-dioxiden |
| US8697255B2 (en) | 2007-07-05 | 2014-04-15 | Basf Se | Organic light-emitting diodes comprising at least one disilyl compound selected from disilylcarbazoles, disilyldibenzofurans, disilyldibenzothiophenes, disilyldibenzopholes, disilyldibenzothiophene S-oxides and disilyldibenzothiophene S,S-dioxides |
| US8384068B2 (en) | 2007-10-02 | 2013-02-26 | Basf Se | Use of acridine derivatives as matrix materials and/or electron blockers in OLEDs |
| DE102008015526B4 (de) | 2008-03-25 | 2021-11-11 | Merck Patent Gmbh | Metallkomplexe |
| DE102008015526A1 (de) | 2008-03-25 | 2009-10-01 | Merck Patent Gmbh | Metallkomplexe |
| US9481826B2 (en) | 2008-06-05 | 2016-11-01 | Merck Patent Gmbh | Electronic device comprising metal complexes |
| US10538698B2 (en) | 2008-06-05 | 2020-01-21 | Merck Patent Gmbh | Electronic device comprising metal complexes |
| DE102008027005A1 (de) | 2008-06-05 | 2009-12-10 | Merck Patent Gmbh | Organische elektronische Vorrichtung enthaltend Metallkomplexe |
| US8859110B2 (en) | 2008-06-20 | 2014-10-14 | Basf Se | Cyclic phosphazene compounds and use thereof in organic light emitting diodes |
| US8618533B2 (en) | 2008-10-07 | 2013-12-31 | Osram Opto Semiconductors Gmbh | Siloles substituted by fused ring systems and use thereof in organic electronics |
| US9169282B2 (en) | 2009-02-02 | 2015-10-27 | Merck Patent Gmbh | Metal complexes |
| WO2010086089A1 (de) | 2009-02-02 | 2010-08-05 | Merck Patent Gmbh | Metallkomplexe |
| DE102009007038A1 (de) | 2009-02-02 | 2010-08-05 | Merck Patent Gmbh | Metallkomplexe |
| WO2010097433A1 (de) | 2009-02-26 | 2010-09-02 | Basf Se | Chinonverbindungen als dotierstoff in der organischen elektronik |
| US11832508B2 (en) | 2009-08-31 | 2023-11-28 | Udc Ireland Limited | Organic electroluminescence device |
| WO2011032626A1 (de) | 2009-09-16 | 2011-03-24 | Merck Patent Gmbh | Metallkomplexe |
| DE102009041414A1 (de) | 2009-09-16 | 2011-03-17 | Merck Patent Gmbh | Metallkomplexe |
| WO2011044988A1 (de) | 2009-10-16 | 2011-04-21 | Merck Patent Gmbh | Metallkomplexe |
| DE102009049587A1 (de) | 2009-10-16 | 2011-04-21 | Merck Patent Gmbh | Metallkomplexe |
| US11189806B2 (en) | 2009-10-28 | 2021-11-30 | Udc Ireland Limited | Heteroleptic carbene complexes and the use thereof in organic electronics |
| US11871654B2 (en) | 2009-10-28 | 2024-01-09 | Udc Ireland Limited | Heteroleptic carbene complexes and the use thereof in organic electronics |
| US11839140B2 (en) | 2009-12-14 | 2023-12-05 | Udc Ireland Limited | Metal complexes comprising diazabenzmidazolocarbene ligands and the use thereof in OLEDS |
| US10916716B2 (en) | 2009-12-14 | 2021-02-09 | Udc Ireland Limited | Metal complexes comprising diazabenzmidazolocarbene ligands and the use thereof in OLEDS |
| US11444254B2 (en) | 2009-12-14 | 2022-09-13 | Udc Ireland Limited | Metal complexes comprising diazabenzmidazolocarbene ligands and the use thereof in OLEDs |
| DE102010020567A1 (de) | 2010-05-14 | 2011-11-17 | Merck Patent Gmbh | Metallkomplexe |
| WO2011141120A1 (de) | 2010-05-14 | 2011-11-17 | Merck Patent Gmbh | Metallkomplexe |
| WO2011157339A1 (de) | 2010-06-15 | 2011-12-22 | Merck Patent Gmbh | Metallkomplexe |
| WO2011157779A1 (en) | 2010-06-18 | 2011-12-22 | Basf Se | Organic electronic devices comprising a layer of a pyridine compound and a 8-hydroxyquinolinolato earth alkaline metal, or alkali metal complex |
| WO2011157790A1 (en) | 2010-06-18 | 2011-12-22 | Basf Se | Organic electronic devices comprising a layer of a dibenzofurane compound and a 8-hydroxyquinolinolato earth alkaline metal, or alkali metal complex |
| US9142792B2 (en) | 2010-06-18 | 2015-09-22 | Basf Se | Organic electronic devices comprising a layer comprising at least one metal organic compound and at least one metal oxide |
| EP3121183A1 (de) | 2010-07-15 | 2017-01-25 | Merck Patent GmbH | Organische komplexe enthaltend metalle |
| DE102010027218A1 (de) | 2010-07-15 | 2012-01-19 | Merck Patent Gmbh | Organische Komplexe enthaltend Metalle |
| EP2896627A1 (de) | 2010-07-15 | 2015-07-22 | Merck Patent GmbH | Organische Komplexe enthaltend Metalle |
| WO2012007103A1 (de) | 2010-07-15 | 2012-01-19 | Merck Patent Gmbh | Metallkomplexe mit organischen liganden und deren verwendung in oleds |
| WO2012007086A1 (de) | 2010-07-16 | 2012-01-19 | Merck Patent Gmbh | Metallkomplexe |
| DE102010027316A1 (de) | 2010-07-16 | 2012-01-19 | Merck Patent Gmbh | Metallkomplexe |
| WO2012007087A1 (de) | 2010-07-16 | 2012-01-19 | Merck Patent Gmbh | Metallkomplexe |
| DE102010027317A1 (de) | 2010-07-16 | 2012-01-19 | Merck Patent Gmbh | Metallkomplexe |
| DE102010046512A1 (de) | 2010-09-24 | 2012-03-29 | Merck Patent Gmbh | Phosphorhaltige Metallkomplexe |
| WO2012038029A1 (de) | 2010-09-24 | 2012-03-29 | Merck Patent Gmbh | Phosphorhaltige metallkomplexe |
| WO2012045710A1 (en) | 2010-10-07 | 2012-04-12 | Basf Se | Phenanthro[9,10-b]furans for electronic applications |
| US9079872B2 (en) | 2010-10-07 | 2015-07-14 | Basf Se | Phenanthro[9, 10-B]furans for electronic applications |
| US8362246B2 (en) | 2010-12-13 | 2013-01-29 | Basf Se | Bispyrimidines for electronic applications |
| WO2012080052A1 (en) | 2010-12-13 | 2012-06-21 | Basf Se | Bispyrimidines for electronic applications |
| US9806270B2 (en) | 2011-03-25 | 2017-10-31 | Udc Ireland Limited | 4H-imidazo[1,2-a]imidazoles for electronic applications |
| US10431750B2 (en) | 2011-03-25 | 2019-10-01 | Udc Ireland Limited | 4H-imidazo[1,2-a]imidazoles for electronic applications |
| WO2012130709A1 (en) | 2011-03-25 | 2012-10-04 | Basf Se | 4h-imidazo[1,2-a]imidazoles for electronic applications |
| EP3034508A1 (de) | 2011-03-25 | 2016-06-22 | Basf Se | 4h-imidazo[1,2-a]imidazole für elektronische anwendungen |
| EP3640252A1 (de) | 2011-03-25 | 2020-04-22 | UDC Ireland Limited | 4h-imidazo[1,2-a]imidazole für elektronische anwendungen |
| DE102012004143A1 (de) | 2011-03-31 | 2012-10-04 | Merck Patent Gmbh | Verfahren zur Herstellung von Iridium-tris(ketoketonaten) |
| WO2012136296A1 (de) | 2011-04-04 | 2012-10-11 | Merck Patent Gmbh | Metallkomplexe |
| US11700769B2 (en) | 2011-08-22 | 2023-07-11 | Udc Ireland Limited | Organic electroluminescent element, compound, and light emitting device, display device and lighting system, using said element |
| US9502664B2 (en) | 2011-11-10 | 2016-11-22 | Udc Ireland Limited | 4H-imidazo[1,2-a]imidazoles for electronic applications |
| WO2013068376A1 (en) | 2011-11-10 | 2013-05-16 | Basf Se | 4h-imidazo[1,2-a]imidazoles for electronic applications |
| WO2013097920A1 (en) | 2011-12-27 | 2013-07-04 | Merck Patent Gmbh | Metal complexes comprising 1,2,3-triazoles |
| EP3232485A1 (de) | 2012-07-10 | 2017-10-18 | UDC Ireland Limited | Benzimidazo[1,2-a]benzimidazol-derivate für elektronische anwendungen |
| US10862051B2 (en) | 2012-07-10 | 2020-12-08 | Udc Ireland Limited | Benzimidazo[1,2-a]benzimidazole derivatives for electronic applications |
| US10243150B2 (en) | 2012-07-10 | 2019-03-26 | Udc Ireland Limited | Benzimidazo[1,2-a]benzimidazole derivatives for electronic applications |
| US9620724B2 (en) | 2012-07-10 | 2017-04-11 | Udc Ireland Limited | Benzimidazo[1,2-A]benzimidazole derivatives for electronic applications |
| WO2014009317A1 (en) | 2012-07-10 | 2014-01-16 | Basf Se | Benzimidazo[1,2-a]benzimidazole derivatives for electronic applications |
| WO2014008982A1 (de) | 2012-07-13 | 2014-01-16 | Merck Patent Gmbh | Metallkomplexe |
| EP3424936A1 (de) | 2012-08-07 | 2019-01-09 | Merck Patent GmbH | Metallkomplexe |
| WO2014023377A2 (de) | 2012-08-07 | 2014-02-13 | Merck Patent Gmbh | Metallkomplexe |
| EP3318566A1 (de) | 2012-09-20 | 2018-05-09 | UDC Ireland Limited | Azadibenzofurane für elektronische anwendungen |
| US10249827B2 (en) | 2012-09-20 | 2019-04-02 | Udc Ireland Limited | Azadibenzofurans for electronic applications |
| WO2014044347A1 (de) | 2012-09-20 | 2014-03-27 | Merck Patent Gmbh | Metallkomplexe |
| WO2014072320A1 (en) | 2012-11-06 | 2014-05-15 | Basf Se | Phenoxasiline based compounds for electronic application |
| US11031559B2 (en) | 2012-11-06 | 2021-06-08 | Udc Ireland Limited | Phenoxasiline based compounds for electronic application |
| US10319917B2 (en) | 2012-11-06 | 2019-06-11 | Udc Ireland Limited | Phenoxasiline based compounds for electronic application |
| WO2014147134A1 (en) | 2013-03-20 | 2014-09-25 | Basf Se | Azabenzimidazole carbene complexes as efficiency booster in oleds |
| EP3608329A1 (de) | 2013-07-02 | 2020-02-12 | UDC Ireland Limited | Monosubstituierte diazabenzimidazolcarben-metall-komplexe zur verwendung in organischen leuchtdioden |
| US11605790B2 (en) | 2013-07-02 | 2023-03-14 | Udc Ireland Limited | Monosubstituted diazabenzimidazole carbene metal complexes for use in organic light emitting diodes |
| WO2015000955A1 (en) | 2013-07-02 | 2015-01-08 | Basf Se | Monosubstituted diazabenzimidazole carbene metal complexes for use in organic light emitting diodes |
| EP3266789A1 (de) | 2013-07-02 | 2018-01-10 | UDC Ireland Limited | Monosubstituierte diazabenzimidazolcarben-metall-komplexe zur verwendung in organischen leuchtdioden |
| WO2015039723A1 (de) * | 2013-09-17 | 2015-03-26 | Merck Patent Gmbh | Polycyclische phenyl-pyridin iridiumkomplexe und derivate davon für oled |
| WO2015063046A1 (en) | 2013-10-31 | 2015-05-07 | Basf Se | Azadibenzothiophenes for electronic applications |
| US10370396B2 (en) | 2014-03-31 | 2019-08-06 | Udc Ireland Limited | Metal complexes, comprising carbene ligands having an O-substituted non-cyclometallated aryl group and their use in organic light emitting diodes |
| US9862739B2 (en) | 2014-03-31 | 2018-01-09 | Udc Ireland Limited | Metal complexes, comprising carbene ligands having an O-substituted non-cyclometalated aryl group and their use in organic light emitting diodes |
| US10118939B2 (en) | 2014-03-31 | 2018-11-06 | Udc Ireland Limited | Metal complexes, comprising carbene ligands having an o-substituted non-cyclometalated aryl group and their use in organic light emitting diodes |
| WO2016016791A1 (en) | 2014-07-28 | 2016-02-04 | Idemitsu Kosan Co., Ltd (Ikc) | 2,9-functionalized benzimidazolo[1,2-a]benzimidazoles as hosts for organic light emitting diodes (oleds) |
| EP2982676A1 (de) | 2014-08-07 | 2016-02-10 | Idemitsu Kosan Co., Ltd. | Benzimidazo[2,1-b]benzoxazole für elektronische Anwendungen |
| EP2993215A1 (de) | 2014-09-04 | 2016-03-09 | Idemitsu Kosan Co., Ltd. | Azabenzimidazo[2,1-a]benzimidazole für elektronische Anwendungen |
| US10333078B2 (en) | 2014-09-26 | 2019-06-25 | Udc Ireland Limited | Fluorescent organic light emitting elements having high efficiency |
| EP3015469A1 (de) | 2014-10-30 | 2016-05-04 | Idemitsu Kosan Co., Ltd. | 5-((benz)imidazol-2-yl)benzimidazo[1,2-a]benzimidazole für elektronische anwendungen |
| WO2016067261A1 (en) | 2014-10-30 | 2016-05-06 | Idemitsu Kosan Co., Ltd. | 5-((benz)imidazol-2-yl)benzimidazo[1,2-a]benzimidazoles for electronic applications |
| WO2016079667A1 (en) | 2014-11-17 | 2016-05-26 | Idemitsu Kosan Co., Ltd. | Indole derivatives for electronic applications |
| WO2016097983A1 (en) | 2014-12-15 | 2016-06-23 | Idemitsu Kosan Co., Ltd. | 1-functionalized dibenzofurans and dibenzothiophenes for organic light emitting diodes (oleds) |
| EP3034506A1 (de) | 2014-12-15 | 2016-06-22 | Idemitsu Kosan Co., Ltd | 4-funktionalisierte Carbazolderivate für elektronische Anwendungen |
| EP3034507A1 (de) | 2014-12-15 | 2016-06-22 | Idemitsu Kosan Co., Ltd | 1-funktionalisierte Dibenzofurane und Dibenzothiophene für organische Leuchtdioden (OLEDS) |
| WO2016124304A1 (de) | 2015-02-03 | 2016-08-11 | Merck Patent Gmbh | Metallkomplexe |
| EP3054498A1 (de) | 2015-02-06 | 2016-08-10 | Idemitsu Kosan Co., Ltd. | Bisimidazodiazozine |
| WO2016125110A1 (en) | 2015-02-06 | 2016-08-11 | Idemitsu Kosan Co., Ltd. | Bisimidazolodiazocines |
| EP3053918A1 (de) | 2015-02-06 | 2016-08-10 | Idemitsu Kosan Co., Ltd | 2-Carbazol substituierte Benzimidazole für elektronische Anwendungen |
| EP3061759A1 (de) | 2015-02-24 | 2016-08-31 | Idemitsu Kosan Co., Ltd | Nitrilsubstituierte dibenzofurane |
| EP3070144A1 (de) | 2015-03-17 | 2016-09-21 | Idemitsu Kosan Co., Ltd. | Siebengliedrige ringverbindungen |
| EP3072943A1 (de) | 2015-03-26 | 2016-09-28 | Idemitsu Kosan Co., Ltd. | Dibenzofuran/carbazol-substituierte benzonitrile |
| WO2016157113A1 (en) | 2015-03-31 | 2016-10-06 | Idemitsu Kosan Co., Ltd. | Benzimidazolo[1,2-a]benzimidazole carrying aryl- or heteroarylnitril groups for organic light emitting diodes |
| EP3075737A1 (de) | 2015-03-31 | 2016-10-05 | Idemitsu Kosan Co., Ltd | Benzimidazolo[1,2-a]benzimidazol mit aryl- oder heteroarylnitrilgruppen für organische leuchtdioden |
| EP3150606A1 (de) | 2015-10-01 | 2017-04-05 | Idemitsu Kosan Co., Ltd. | Benzimidazolo[1,2-a]benzimidazole mit benzofuran- oder benzothiophen- gruppen für organische licht emittierende dioden. |
| WO2017056053A1 (en) | 2015-10-01 | 2017-04-06 | Idemitsu Kosan Co., Ltd. | Benzimidazolo[1,2-a]benzimidazole carrying benzimidazolo[1,2-a]benzimidazolyl groups, carbazolyl groups, benzofurane groups or benzothiophene groups for organic light emitting diodes |
| WO2017056055A1 (en) | 2015-10-01 | 2017-04-06 | Idemitsu Kosan Co., Ltd. | Benzimidazolo[1,2-a]benzimidazole carrying triazine groups for organic light emitting diodes |
| EP3150604A1 (de) | 2015-10-01 | 2017-04-05 | Idemitsu Kosan Co., Ltd. | Benzimidazolo[1,2-a]benzimidazol mit benzimidazolo[1,2-a]benzimidazolylgruppen, carbazolylgruppen, benzofurangruppen oder benzothiophengruppen für organische leuchtdioden |
| WO2017056052A1 (en) | 2015-10-01 | 2017-04-06 | Idemitsu Kosan Co., Ltd. | Benzimidazolo[1,2-a]benzimidazole carrying benzimidazolo[1,2-a]benzimidazolyl groups, carbazolyl groups, benzofurane groups or benzothiophene groups for organic light emitting diodes |
| WO2017078182A1 (en) | 2015-11-04 | 2017-05-11 | Idemitsu Kosan Co., Ltd. | Benzimidazole fused heteroaryls |
| WO2017093958A1 (en) | 2015-12-04 | 2017-06-08 | Idemitsu Kosan Co., Ltd. | Benzimidazolo[1,2-a]benzimidazole derivatives for organic light emitting diodes |
| WO2017109727A1 (en) | 2015-12-21 | 2017-06-29 | Idemitsu Kosan Co., Ltd. | Hetero-condensed phenylquinazolines and their use in electronic devices |
| WO2017109722A1 (en) | 2015-12-21 | 2017-06-29 | Idemitsu Kosan Co., Ltd. | Nitrogen-containing heterocyclic compounds and organic electroluminescence devices containing them |
| WO2017178864A1 (en) | 2016-04-12 | 2017-10-19 | Idemitsu Kosan Co., Ltd. | Seven-membered ring compounds |
| WO2017221999A1 (en) | 2016-06-22 | 2017-12-28 | Idemitsu Kosan Co., Ltd. | Specifically substituted benzofuro- and benzothienoquinolines for organic light emitting diodes |
| WO2018001990A1 (de) | 2016-06-30 | 2018-01-04 | Merck Patent Gmbh | Verfahren zur auftrennung von enantiomerenmischungen von metallkomplexen |
| WO2018011186A1 (de) | 2016-07-14 | 2018-01-18 | Merck Patent Gmbh | Metallkomplexe |
| WO2018019687A1 (de) | 2016-07-25 | 2018-02-01 | Merck Patent Gmbh | Di- und oligonukleare metallkomplexe mit tripodalen bidentaten teilliganden sowie deren verwendung in elektronischen vorrichtungen |
| WO2018019688A1 (de) | 2016-07-25 | 2018-02-01 | Merck Patent Gmbh | Metallkomplexe für den einsatz als emitter in organischen elektrolumineszenzvorrichtungen |
| WO2018041769A1 (de) | 2016-08-30 | 2018-03-08 | Merck Patent Gmbh | Bl- und trinukleare metallkomplexe aufgebaut aus zwei miteinander verknüpften tripodalen hexadentaten liganden zur verwendung in elektrolumineszenzvorrichtungen |
| WO2018054798A1 (de) | 2016-09-21 | 2018-03-29 | Merck Patent Gmbh | Binukleare metallkomplexe für den einsatz als emitter in organischen elektrolumineszenzvorrichtungen |
| WO2018069197A1 (de) | 2016-10-12 | 2018-04-19 | Merck Patent Gmbh | Metallkomplexe |
| WO2018069196A1 (de) | 2016-10-12 | 2018-04-19 | Merck Patent Gmbh | Binukleare metallkomplexe sowie elektronische vorrichtungen, insbesondere organische elektrolumineszenzvorrichtungen, enthaltend diese metallkomplexe |
| WO2018069273A1 (de) | 2016-10-13 | 2018-04-19 | Merck Patent Gmbh | Metallkomplexe |
| WO2018077769A1 (de) | 2016-10-25 | 2018-05-03 | Merck Patent Gmbh | Metallkomplexe |
| US11267835B2 (en) | 2017-02-14 | 2022-03-08 | Merck Patent Gmbh | Process for preparing ortho-metallated metal compounds |
| WO2018177981A1 (de) | 2017-03-29 | 2018-10-04 | Merck Patent Gmbh | Aromatische verbindungen |
| WO2019020538A1 (de) | 2017-07-25 | 2019-01-31 | Merck Patent Gmbh | Metallkomplexe |
| EP3466954A1 (de) | 2017-10-04 | 2019-04-10 | Idemitsu Kosan Co., Ltd. | Mit einem heteroatom verbrückte kondensierte phenylchinazoline |
| WO2019158453A1 (de) | 2018-02-13 | 2019-08-22 | Merck Patent Gmbh | Metallkomplexe |
| WO2019179909A1 (de) | 2018-03-19 | 2019-09-26 | Merck Patent Gmbh | Metallkomplexe |
| EP3604477A1 (de) | 2018-07-30 | 2020-02-05 | Idemitsu Kosan Co., Ltd. | Polycyclische verbindung, organische elektrolumineszenzvorrichtung und elektronische vorrichtung |
| WO2020026133A1 (en) | 2018-07-30 | 2020-02-06 | Idemitsu Kosan Co., Ltd. | Polycyclic compound, organic electroluminescence device, and electronic device |
| EP3608319A1 (de) | 2018-08-07 | 2020-02-12 | Idemitsu Kosan Co., Ltd. | Kondensierte azazyklen als organische lichtemittierende vorrichtungen und materialien zur verwendung darin |
| WO2020165064A1 (de) | 2019-02-11 | 2020-08-20 | Merck Patent Gmbh | Mononukleare iridiumkomplexe mit drei ortho-metallierten bidentaten liganden und optischer orientierungsanisotropie |
| WO2020212296A1 (de) | 2019-04-15 | 2020-10-22 | Merck Patent Gmbh | Metallkomplexe |
| WO2021110720A1 (de) | 2019-12-04 | 2021-06-10 | Merck Patent Gmbh | Metallkomplexe |
| WO2022069380A1 (de) | 2020-09-29 | 2022-04-07 | Merck Patent Gmbh | Mononukleare tripodale hexadentate iridium komplexe zur verwendung in oleds |
| EP4311849A1 (de) | 2022-07-27 | 2024-01-31 | UDC Ireland Limited | Metallkomplexe |
Also Published As
| Publication number | Publication date |
|---|---|
| KR100934147B1 (ko) | 2009-12-29 |
| US20040077862A1 (en) | 2004-04-22 |
| US7423151B2 (en) | 2008-09-09 |
| CN1527835A (zh) | 2004-09-08 |
| CN100362004C (zh) | 2008-01-16 |
| CN100528885C (zh) | 2009-08-19 |
| JP2004526700A (ja) | 2004-09-02 |
| EP1366054B1 (de) | 2005-04-06 |
| CN1781926A (zh) | 2006-06-07 |
| JP3984167B2 (ja) | 2007-10-03 |
| KR20030077599A (ko) | 2003-10-01 |
| DE50202715D1 (de) | 2005-05-12 |
| US7084273B2 (en) | 2006-08-01 |
| US20060252936A1 (en) | 2006-11-09 |
| EP1366054A1 (de) | 2003-12-03 |
| DE10104426A1 (de) | 2002-08-08 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP1366054B1 (de) | Verfahren zur herstellung von hochreinen, tris-ortho-metallierten organo-iridium-verbindungen | |
| EP1611146B1 (de) | Verfahren zur herstellung von hochreinen organo-iridium-verbindungen | |
| EP1534722B1 (de) | Rhodium-und iridium-komplexe | |
| DE10223337A1 (de) | Verfahren zur Herstellung von hochreinen, tris-orthometallierten Organo-Iridium-Verbindungen | |
| EP1749014B1 (de) | Metallkomplexe | |
| EP1678190A1 (de) | Metallkomplexe mit bipodalen liganden | |
| EP1558619B1 (de) | Rhodium und iridium-komplexe | |
| EP1504015A1 (de) | Rhodium- und iridium-komplexe | |
| WO2002081488A1 (de) | Rhodium-und iridium-komplexe | |
| EP1622919B1 (de) | Verfahren zur herstellung von tris-ortho-metallierten organometallkomplexen und verwendung solcher komplexe in oleds | |
| EP1776374B1 (de) | Verfahren zur herstellung von iridium(iii)ketoketonaten | |
| EP1636244B1 (de) | Verfahren zur herstellung von hochreinen, tris- und bis-ortho-metallierten organometall-verbindungen | |
| EP1448577B1 (de) | Rhodium- und iridium-komplexe |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): CN JP KR US |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): AT BE CH CY DE DK ES FI FR GB GR IE IT LU MC NL PT SE TR |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 2002710817 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 028042123 Country of ref document: CN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1020037010096 Country of ref document: KR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2002561478 Country of ref document: JP |
|
| WWP | Wipo information: published in national office |
Ref document number: 1020037010096 Country of ref document: KR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 10470811 Country of ref document: US |
|
| WWP | Wipo information: published in national office |
Ref document number: 2002710817 Country of ref document: EP |
|
| WWG | Wipo information: grant in national office |
Ref document number: 2002710817 Country of ref document: EP |