WO2001034594A1 - Dipeptidyl peptidase iv inhibitors and methods of making and using dipeptidyl peptidase iv inhibitors - Google Patents
Dipeptidyl peptidase iv inhibitors and methods of making and using dipeptidyl peptidase iv inhibitors Download PDFInfo
- Publication number
- WO2001034594A1 WO2001034594A1 PCT/US2000/030836 US0030836W WO0134594A1 WO 2001034594 A1 WO2001034594 A1 WO 2001034594A1 US 0030836 W US0030836 W US 0030836W WO 0134594 A1 WO0134594 A1 WO 0134594A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- amino
- branched
- inhibitor
- cycloalkyl
- group
- Prior art date
Links
- 0 CC(*)CC(C(N(CNC1)C1C(O)=*)=S)Nc1ccccc1 Chemical compound CC(*)CC(C(N(CNC1)C1C(O)=*)=S)Nc1ccccc1 0.000 description 8
- DXLCUOPSMUWYFG-JTQLQIEISA-N CCC(C)(C)NCC(N(CCC1)[C@@H]1C#N)=O Chemical compound CCC(C)(C)NCC(N(CCC1)[C@@H]1C#N)=O DXLCUOPSMUWYFG-JTQLQIEISA-N 0.000 description 1
- IOAMDAFPCXAGIS-UHFFFAOYSA-N CCNC(C)C(N(CC(O)=O)Cc1ccccc1)=O Chemical compound CCNC(C)C(N(CC(O)=O)Cc1ccccc1)=O IOAMDAFPCXAGIS-UHFFFAOYSA-N 0.000 description 1
- MPQZBMGBCWKCQH-AWEZNQCLSA-N COc1ccc(CCNCC(N(CCC2)[C@@H]2C#N)=O)cc1 Chemical compound COc1ccc(CCNCC(N(CCC2)[C@@H]2C#N)=O)cc1 MPQZBMGBCWKCQH-AWEZNQCLSA-N 0.000 description 1
- HOUBYLHOXLBMIT-HNNXBMFYSA-N N#C[C@H](CCC1)N1C(CNC(C1)Cc2c1cccc2)=O Chemical compound N#C[C@H](CCC1)N1C(CNC(C1)Cc2c1cccc2)=O HOUBYLHOXLBMIT-HNNXBMFYSA-N 0.000 description 1
- DKVVXAOVBVZNMB-HNNXBMFYSA-N N#C[C@H](CCC1)N1C(CNCCCc1ccccc1)=O Chemical compound N#C[C@H](CCC1)N1C(CNCCCc1ccccc1)=O DKVVXAOVBVZNMB-HNNXBMFYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D257/00—Heterocyclic compounds containing rings having four nitrogen atoms as the only ring hetero atoms
- C07D257/02—Heterocyclic compounds containing rings having four nitrogen atoms as the only ring hetero atoms not condensed with other rings
- C07D257/04—Five-membered rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/08—Drugs for disorders of the urinary system of the prostate
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/06—Antimigraine agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/14—Drugs for disorders of the nervous system for treating abnormal movements, e.g. chorea, dyskinesia
- A61P25/16—Anti-Parkinson drugs
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C229/00—Compounds containing amino and carboxyl groups bound to the same carbon skeleton
- C07C229/46—Compounds containing amino and carboxyl groups bound to the same carbon skeleton having amino or carboxyl groups bound to carbon atoms of rings other than six-membered aromatic rings of the same carbon skeleton
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C237/00—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by amino groups
- C07C237/02—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by amino groups having the carbon atoms of the carboxamide groups bound to acyclic carbon atoms of the carbon skeleton
- C07C237/04—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by amino groups having the carbon atoms of the carboxamide groups bound to acyclic carbon atoms of the carbon skeleton the carbon skeleton being acyclic and saturated
- C07C237/12—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by amino groups having the carbon atoms of the carboxamide groups bound to acyclic carbon atoms of the carbon skeleton the carbon skeleton being acyclic and saturated having the nitrogen atom of at least one of the carboxamide groups bound to an acyclic carbon atom of a hydrocarbon radical substituted by carboxyl groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C255/00—Carboxylic acid nitriles
- C07C255/45—Carboxylic acid nitriles having cyano groups bound to carbon atoms of rings other than six-membered aromatic rings
- C07C255/46—Carboxylic acid nitriles having cyano groups bound to carbon atoms of rings other than six-membered aromatic rings to carbon atoms of non-condensed rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C327/00—Thiocarboxylic acids
- C07C327/38—Amides of thiocarboxylic acids
- C07C327/40—Amides of thiocarboxylic acids having carbon atoms of thiocarboxamide groups bound to hydrogen atoms or to acyclic carbon atoms
- C07C327/42—Amides of thiocarboxylic acids having carbon atoms of thiocarboxamide groups bound to hydrogen atoms or to acyclic carbon atoms to hydrogen atoms or to carbon atoms of a saturated carbon skeleton
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D207/00—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D207/02—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D207/04—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members
- C07D207/10—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D207/16—Carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D233/00—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings
- C07D233/02—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings having no double bonds between ring members or between ring members and non-ring members
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D241/00—Heterocyclic compounds containing 1,4-diazine or hydrogenated 1,4-diazine rings
- C07D241/02—Heterocyclic compounds containing 1,4-diazine or hydrogenated 1,4-diazine rings not condensed with other rings
- C07D241/04—Heterocyclic compounds containing 1,4-diazine or hydrogenated 1,4-diazine rings not condensed with other rings having no double bonds between ring members or between ring members and non-ring members
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D263/00—Heterocyclic compounds containing 1,3-oxazole or hydrogenated 1,3-oxazole rings
- C07D263/02—Heterocyclic compounds containing 1,3-oxazole or hydrogenated 1,3-oxazole rings not condensed with other rings
- C07D263/04—Heterocyclic compounds containing 1,3-oxazole or hydrogenated 1,3-oxazole rings not condensed with other rings having no double bonds between ring members or between ring members and non-ring members
- C07D263/06—Heterocyclic compounds containing 1,3-oxazole or hydrogenated 1,3-oxazole rings not condensed with other rings having no double bonds between ring members or between ring members and non-ring members with hydrocarbon radicals, substituted by oxygen atoms, attached to ring carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D277/00—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings
- C07D277/02—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings not condensed with other rings
- C07D277/04—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings not condensed with other rings having no double bonds between ring members or between ring members and non-ring members
- C07D277/06—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings not condensed with other rings having no double bonds between ring members or between ring members and non-ring members with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D279/00—Heterocyclic compounds containing six-membered rings having one nitrogen atom and one sulfur atom as the only ring hetero atoms
- C07D279/10—1,4-Thiazines; Hydrogenated 1,4-thiazines
- C07D279/12—1,4-Thiazines; Hydrogenated 1,4-thiazines not condensed with other rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D307/00—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom
- C07D307/02—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom not condensed with other rings
- C07D307/04—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom not condensed with other rings having no double bonds between ring members or between ring members and non-ring members
- C07D307/18—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom not condensed with other rings having no double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D307/24—Carbon atoms having three bonds to hetero atoms with at the most one bond to halogen
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/04—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings
- C07D413/04—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings
- C07D417/04—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F9/00—Compounds containing elements of Groups 5 or 15 of the Periodic System
- C07F9/02—Phosphorus compounds
- C07F9/28—Phosphorus compounds with one or more P—C bonds
- C07F9/38—Phosphonic acids RP(=O)(OH)2; Thiophosphonic acids, i.e. RP(=X)(XH)2 (X = S, Se)
- C07F9/40—Esters thereof
- C07F9/4003—Esters thereof the acid moiety containing a substituent or a structure which is considered as characteristic
- C07F9/4006—Esters of acyclic acids which can have further substituents on alkyl
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F9/00—Compounds containing elements of Groups 5 or 15 of the Periodic System
- C07F9/02—Phosphorus compounds
- C07F9/28—Phosphorus compounds with one or more P—C bonds
- C07F9/48—Phosphonous acids R—P(OH)2; Thiophosphonous acids including RHP(=O)(OH); Derivatives thereof
- C07F9/4808—Phosphonous acids R—P(OH)2; Thiophosphonous acids including RHP(=O)(OH); Derivatives thereof the acid moiety containing a substituent or structure which is considered as characteristic
- C07F9/4833—Cycloaliphatic acids or derivatives
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F9/00—Compounds containing elements of Groups 5 or 15 of the Periodic System
- C07F9/02—Phosphorus compounds
- C07F9/547—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom
- C07F9/553—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom having one nitrogen atom as the only ring hetero atom
- C07F9/572—Five-membered rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C2601/00—Systems containing only non-condensed rings
- C07C2601/06—Systems containing only non-condensed rings with a five-membered ring
- C07C2601/08—Systems containing only non-condensed rings with a five-membered ring the ring being saturated
Definitions
- the present invention relates to new and improved inhibitors of Dipepudyl Peptidase IV ("DPP IV"), and new and improved treatment methods and related uses.
- DPP IV inhibitors according to the invention are useful for treating a wide variety of diseases and other abnormal conditions, including diseases impacting the central nervous system.
- Dipeptidyl peptidase IV is a membrane-bound peptidase involved in the release of N-terminal dipeptides from proteins and other types or forms of peptides.
- the enzyme is a type ⁇ membrane serine peptidase, and has a preference for removing proline-containing dipeptides from the N-terminus of the protein or peptide.
- the enzyme contains 767 amino acids, and has been found in the kidney, epithelial cells, endothelial cells, small intestine, prostrate, seminal plasma and the brain.
- the physiological roles of DPP IV have not been completely elucidated. It has been thought that DPP IV plays a role in the cleavage of various cytokines, growth factors and neuropeptides.
- the enzyme also can cleave neuropeptides such as substance P and neuropeptide Y.
- DPP IV is involved in cell adhesion and with the T-cell activation marker CD26.
- DPP IV has been implicated in disease states such as HTV infection, diabetes, arthritis and certain cancers.
- a DPP IV presence has been implicated in prostate and lung cancer, and DPP IV also has been found in patients having benign prostate hyperplasia.
- DPP IV also is being investigated for its role in type II diabetes because the glucagon-like peptide (GLP-1) can be a substrate for DPP IV cleavage, and some DPP IV inhibitors have demonstrated efficacy in animal models for diabetes.
- GLP-1 glucagon-like peptide
- DPP IV has been implicated in HTV infection due to its association with CD 26. DPP IV also has been identified as a "research front" in an article about Alzheimer's disease. Shvaloff et al. , DIALOG FILE NO. 05335738/5.
- DPP IV inhibition has been shown to increase release of TGF- ⁇ , a protein having neuroprotective properties. DPP IV inhibition itself, however, has not been implicated in a neuroprotective context.
- DPP IV inhibition has been studied in the treatment of autoimmune diseases such as diabetes, arthritis and multiple sclerosis (a demyelination disease of the peripheral nerves). See PCT publications WO 97/40832 and WO 98/19998. Additionally, PCT publication WO 94/03055 discusses increasing production of hematopoietic cells with DPP IV inhibitors. PCT publication WO 95/11689 discloses the use of DPP IV inhibitors to block the entry of HIV into cells. U.S. Patent No. 5,543,396 discloses the use of inhibitors (certain proline phosphonate derivatives) to treat tumor invasion.
- PCT publication WO 95/34538 mentions the use of certain serine protease inhibitors (such as certain DPP IV and PEP inhibitors) to treat peripheral neurological/autoimmune diseases like multiple sclerosis.
- DPP IV inhibitors based upon molecules that bear a resemblance to proline have been investigated in the field.
- PCT publication WO 95/11689 discloses c - amino boronic acid analogs of proline.
- PCT publication WO 98/19998 discloses N- substituted 2-cyanopyrrolidines as DPP IV inhibitors.
- PCT publication WO 95/34538 also discloses various proline containing compounds. Alexander et al., BIOSIS NO.
- U.S. Patent Nos. 6,011,155; 6,110,949; and 6,124,305 discloses various N-substituted cyanopyrrolidines and cyanothiazolidines to inhibit DPP IV for the treatment of diabetes, and "conditions mediated by dipeptidyl peptidase- V inhibition. " The field, however, lacks appreciation of the usefulness of DPP IV inhibition for treating disease states, injuries and other abnormal conditions involving the central nervous system and other parts of the body, such as in the treatment of prostate.
- inhibitors of dipeptidyl peptidase IV can include a proline mimetic and preferably possess an IC J0 of no more than about 1 ⁇ m, preferably no more than 100 nm, and have molecular weights of no more than 700, preferably no more than about 500.
- the inhibitors are reversible.
- the inhibitors preferably are sufficiently neutral and non-polar such that they can cross the blood-brain barrier via passive diffusion. In many cases, inhibitors that cannot cross by passive diffusion instead cross by active transport.
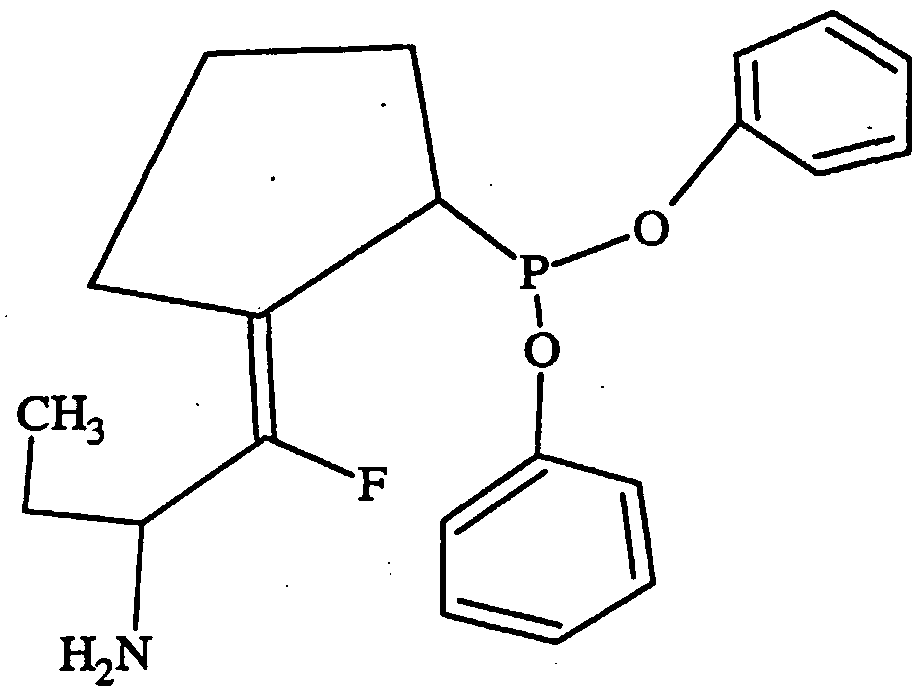
- Inhibitors for use according to the invention include c-KPG and inhibitors according to Core Structures I, II , HI or IV, as shown below.
- reversible inhibitors of dipeptidyl peptidase IV wherein the inhibitor is preferably reversible and preferably has a core structure of selected from the group consisting of Core Structure I, Core Structure II, Core Structure HI and Core Structure IV.
- a given core structure can have functional and substitution groups, such as X, X,, A, Z and R, wherein X (if present) is CR2R3, O, S, or NR4; X, (if present) is CR2R3, O, S, or NR4 with the optional proviso that X and Xj cannot both be a heteroatom; A is H, COOH, or isosteres of carboxylic acids, such as one selected from the group consisting of CN, SO 3 H, CONOH, PO 3 R5R6, SO 2 NHR7, tetrazole, amides, esters, and acid anhydrides; Z (if present) is O or S; and the various R groups that are present are independently selected from the group of functional groups consisting of H, C r C 9 branched or straight chain alkyl, C 2 -C 9 branched or straight chain alkenyl, C 3 -C 8 cycloalkyl, C 5 -C 7 cycloalkenyl,
- the inhibitors for use in such methods preferably should be reversible and preferably be able to cross the blood-brain barrier in amounts sufficient to treat the disorder.
- the compounds according to the invention can be administered concurrently or sequentially with other compounds.
- different compounds according to the invention e.g. , different compounds of one core structure group or compounds of two or more of the core structure groups
- Uses of the compounds disclosed herein are provided (1) for treating disorders of the central nervous system and (2) for preparing compositions, formulations and medicaments for treating disorders of the central nervous system.
- the inhibitors for use in such methods should be reversible and be able to penetrate or act upon the prostate.
- the compounds according to the invention can be administered concurrently or sequentially with other compounds.
- different compounds according to the invention e.g. , different compounds of one core structure group or compounds of two or more of the core structure groups
- Uses of the compounds disclosed herein are provided (1) for treating disorders of the prostate and (2) for preparing compositions, formulations and medicaments for treating disorders of the prostate.
- FIGURE 1 graphically depicts an assay employing organotypic spinal motor neurons and threohydroxyaspartate ("THA"). Exposure of neurons with THA alone resulted in death of 55-60% of the neurons. When the neurons were exposed to THA in combination with 10 ⁇ M c-KPG, the c-KPG spared greater than 50% of the neurons that would have otherwise been killed.
- THA threohydroxyaspartate
- the present invention provides DPP IV inhibitors that are useful for treating various disorders, including those of the central nervous system, among others.
- the DPP IV inhibitors are pyrrolidine-based compounds, and more preferably constitute or include proline or proline mimetics.
- the compounds according to the present invention preferably have sufficient stability, potency, selectivity, solubility and availability to be safe and effective in treating diseases, injuries and other abnormal conditions or insults to the central nervous system, the peripheral nerves and the prostate, for example.
- treat in its various grammatical forms as used in relation to the present invention refers to preventing, curing, reversing, attenuating, alleviating, minimizing, suppressing, ameliorating or halting the deleterious effects of a disease state, disease progression, injury, wound, ischemia, disease causative agent (e.g. , bacteria, protozoans, parasites, fungi, viruses, viroids and/or prions), surgical procedure or other abnormal or detrimental condition (all of which are collectively referred to as "disorders,” as will be appreciated by the person of skill in the art).
- a “therapeutically effective amount” of an inhibitor according to the invention is an amount that can achieve effective treatment, and such amounts can be determined in accordance with the present teachings.
- DPP IV exhibits a preference for causing the removal of proline-containing dipeptides from the N-terminus of a protein or a peptide. Accordingly, proline has a structure that likely is recognized by or acted upon by the active site of DPP IV. Proline is unique among the 20 naturally-occurring amino acids in that it contains a cyclic secondary amino group, which as a result causes it to create interruptions in alpha- helical structures in proteins or peptides.
- the DPP IV inhibitors according to the present invention can constitute or include proline or proline-like moieties, often referred to as "proline mimetics.”
- a proline mimetic is a structure that sufficiently resembles proline such that its charge, polarity, shape and size are sufficiently duplicative of proline so as to participate in many of the molecular interactions involving proline.
- a molecule or other compound that includes a proline moiety can itself be considered a proline mimetic. Accordingly, molecules that constitute or include proline or proline mimetics can interact with the natural interaction partners of proline, such as DPP IV.
- a DPP IV inhibitor has the same or greater affinity for DPP IV than does the natural substrate of DPP IV, such as a protein containing a proline residue at its N-terminal end.
- the inhibitor will have an equal or greater affinity to permit it to more effectively compete for the active site of DPP IV.
- Inhibitors with lower affinities are still within the scope of the invention, and effective competition, and thus inhibition, can be ensured through dosing considerations.
- the DPP IV inhibitor is used to treat disorders of the prostate, including, but not limited to, prostate cancer and post- prostatectomy nerve recovery.
- erectile and voiding disorders are extremely common clinical conditions that result from diseases, injuries and trauma including complications associated with pelvic surgery. It is believed that local nerve injury during major pelvic surgeries account for complications such as erectile dysfunction and urinary incontinence. These complications might be caused by the trauma or the injury of the nerves (e.g. cavernous nerve) innervating the area during the surgery. Appropriate administration of a DPP IV inhibitors prior to, during or after surgery may be effective in blocking the nerve degeneration caused by pelvic surgery.
- the inhibitor of the invention can be administered in the manner used with other prostate therapeutics, and can be combined with other products or methodologies for treating the prostate.
- the DPP IV inhibitor can be used to treat disorders of the central nervous system (CNS) and the peripheral nerves.
- CNS central nervous system
- the DPP IV inhibitors according to the present invention can be used to treat CNS maladies such as strokes, tumors, ischemia, Parkinson's disease, memory loss, hearing loss, vision loss, migraines, brain injury, spinal cord injury, Alzheimer's disease and amyotrophic lateral sclerosis (which has a CNS component).
- the DPP IV inhibitors can be used to treat disorders having a more peripheral nature, including multiple sclerosis and diabetic neuropathy.
- the blood-brain barrier prevents many compounds in the circulation from crossing to the brain.
- the brain is a complex biological structure that is susceptible to a variety of toxins. Additionally, being that the brain is composed primarily of nerves and related tissues, the brain lacks the natural regenerative capabilities of other organs and tissues. For example, the skin has extensive regeneration and restorative capabilities, and thus can withstand encounters with toxins and other physical insults, which it can be expected to encounter in daily life.
- the brain itself is quite susceptible to toxins, and thus it is thought that the blood-brain barrier was an evolutionary development to protect the integrity of the brain.
- the blood-brain barrier also can prevent the entry of beneficial compounds, such as drugs, that are needed to treat a disease, injury or other abnormal condition. Accordingly, the blood-brain barrier can be a complicating factor in developing therapeutics for the CNS.
- a compound that crosses the blood-brain barrier via passive diffusion should have a log P between about 1 and about 4.
- the log D which takes into consideration the charge of the compound.
- polar and charged compounds are less amenable to crossing the blood-brain barrier by passive diffusion. Accordingly, a log D greater than about -2 is preferred.
- the concepts of log P and log D are discussed in Waterbeemed, STRUCTURAL-PROPERTY CORRELATIONS IN DRUG RESEARCH (Academic Press).
- the compound preferably has a molecular weight of about 700 or less, preferably about 500 or less.
- a compound that is to cross the blood-brain barrier by passive diffusion should be "sufficiently neutral and non-polar" for its size that it can cross the blood-brain barrier in a therapeutically effective amount
- potency of the compound as an inhibitor Besides efficiency of a compound in crossing the blood-brain barrier, another important consideration is the potency of the compound as an inhibitor.
- potent inhibitors can have a lower efficiency in crossing the blood-brain barrier, but nevertheless can be effective due to their higher potencies. Conversely, a less potent inhibitor may require greater efficiency in crossing the blood-brain barrier in order to have a beneficial effect.
- a therapeutically effective amount for treating a CNS disorder depends upon the potency of the inhibitor and its efficiency in crossing the blood-brain barrier or the administration route and approach employed to circumvent the blood-brain barrier.
- the DPP IV inhibitors preferably have an IC 50 (for inhibition concentration where 50% of DPP IV is inhibited) value of less than about 1 ⁇ m, and preferably less than 100 ran.
- IC 50 for inhibition concentration where 50% of DPP IV is inhibited
- DPP IV inhibitors can have higher IC 50 values as long as their efficiency in crossing the blood-brain barrier is sufficient to treat the disease, injury or other abnormal condition.
- the DPP IV inhibitor according to the invention is a reversible inhibitor. That is, the DPP IV inhibitor should be able to interact with the inhibitor without becoming permanently bound thereto in a manner that would denature or inactivate the DPP IV enzyme. The need for reversibility is due to the fact that DPP IV is a naturally- occurring enzyme that has normal physiologic functions.
- An irreversible inhibitor can effectively eliminate functions of the enzyme, and thus result in cessation of normal physiologic processes.
- the present invention utilizes the inhibition of DPP IV in certain contexts, such as in treating an ischemic event, for definite periods of time, such as during and after reperfusion in the ischemic area.
- a reversible inhibitor would permit inhibited DPP IV molecules to resume normal function once the need for inhibition is gone.
- the compounds according to the invention can be administered by a variety of systemic and CNS-targeted routes.
- intra-arterial, intravenous intraventricular, intracavitary and intracranial administration routes can be employed.
- Exemplary injection modalities can be by way of bolus, periodic injection and/or constant infusion.
- the following routes can be employed for the compounds according to the invention, including parenteral, oral, nasal, inhalation spray, buccally, topically, transdermal, rectal, vaginal, via implanted reservoir or other routes available to the skilled person.
- parenteral as used herein includes subcutaneous, intravenous, intramuscular, intraperitoneal, intrathecal, intraventricular, intrasternal, intracranial or intraosseous injection and infusion techniques.
- the compounds of the present invention preferably penetrate the blood-train barrier when peripherally administered. Compounds which cannot sufficiently penetrate the blood-brain barrier can be effectively administered by an intraventricular route. It also is important to note that during the active phase of certain CNS disorders, blood-brain lineage is known to occur and will permit entry of the compounds of the invention to the central nervous system. Moreover, there are several other techniques that either physically break through the blood-brain barrier or circumvent it to deliver therapeutic agents. Examples of these techniques include intrathecal injections, surgical implants, and osmotic techniques. Invasive techniques often are employed, particularly direct administration to damaged neuronal tissue. One or more of the above can be employed according to the invention.
- One embodiment for the administration of the compounds of the invention is by intrathecal injection, i.e., directly into the cerebrospinal fluid by puncturing the membranes surrounding the central nervous system is usually by lumbar puncture. Sustained dosages of agents directly into the cerebrospinal fluid can be attained by the use of infusion pumps that are implanted surgically.
- Another embodiment for the administration of the compounds of the invention is by injection directly into the lumbar cerebrospinal fluid (intrathecally) or by injection intravenously.
- the compounds according to the invention can be formulated with pharmaceutically- acceptable carriers and diluents, and can be used with methods and uses according to the invention.
- the formulation will depend upon the disease state being treated and the administration route. See, for example, U.S. Patent No. 5,874,449, which is incorporated by reference.
- Pharmaceutically acceptable carriers include aqueous solutions, non-toxic excipients, including salts, preservatives, buffers, such as phosphate buffers, and the like, as described in UNITED STATES PHARMACOPEIA AND NATIONAL FORMULARY (USP 24-NF 19); REMINGTON'S PHARMACEUTICAL SCIENCES; HANDBOOK ON PHARMACEUTICAL EXCIPIENTS (2d edition, Wade and Weller eds. 1994), the each of which are hereby incorporated by reference.
- non-aqueous solvents are propylene glycol, polyethylene glycol, vegetable oil and injectable organic esters such as ethyloleate.
- Aqueous carriers include water, alcoholic/aqueous solutions, saline solutions, parenteral vehicles, such as sodium chloride and Ringer's dextrose.
- Intravenous vehicles include fluid and nutrient replenishers.
- Preservatives include antimicrobials, anti-oxidants, chelating agents and inert gases.
- the pH and exact concentration of the various components of the binding composition are adjusted according to routine skills in the art. See GOODMAN AND GILMAN'S THE PHARMACOLOGICAL BASIS FOR THERAPEUTICS (9th edition), the contents of which are hereby incorporated by reference.
- Exemplary approaches include those where the compounds are to be administered in the form of sterile injectable preparations, for example, as sterile injectable aqueous or oleaginous suspensions. These suspensions can be formulated according to techniques known in the art using suitable dispersing or wetting agents and suspending agents.
- the sterile injectable preparations may also be sterile injectable solutions or suspensions in non- toxic parenterally-acceptable diluents or solvents, for example, as solutions in 1,3- butanediol.
- acceptable vehicles and solvents that may be employed are water, Ringer's solution and isotonic sodium chloride solution.
- sterile fixed oils are conventionally employed as solvents or suspending mediums.
- any blank fixed oil such as a synthetic mono- or di-glyceride may be employed.
- Fatty acids such as a oleic acid and its glyceride derivatives, including olive oil and castor oil, especially in their polyoxyethylated forms, are useful in the preparation of injectables.
- These oil solutions or suspensions may also contain long-chain alcohol diluents or dispersants.
- the compounds may be administered orally in the form of capsules, tablets, aqueous suspensions or solutions; Tablets may contain carriers such as lactose and corn starch, and/or lubricating agents such as magnesium stearate.
- Capsules may contain diluents including lactose and dried corn starch.
- Aqueous suspensions may contain emulsifying and suspending agents combined with the active ingredient.
- the oral dosage forms may further contain sweetening and/or flavoring and/or coloring agents.
- compositions can be prepared by mixing the drug with suitable non-irritating excipients which are solid at room temperature, but liquid at rectal temperature such that they will melt in the rectum to release the drug.
- suitable non-irritating excipients include cocoa butter, beeswax and polyethylene glycols.
- the compounds may be administered topically, especially when the conditions addressed for treatment involve areas or organs readily accessible by topical application, including neurological disorders of the eye, the skin or the lower intestinal tract.
- the compounds can be formulated as micronized suspensions in isotonic, pH adjusted sterile saline or, preferably, as a solution in isotonic, pH adjusted sterile saline, either with or without a preservative such as benzylalkonium chloride.
- the compounds may be formulated into ointments, such as petrolatum.
- the compounds can be formulated into suitable ointments containing the compounds suspended or dissolved in, for example, mixtures with one or more of the following: mineral oil, liquid petrolatum, white petrolatum, propylene glycol, polyoxyethylene polyoxypropylene compound, emulsifying wax and water.
- the compounds can be formulated into suitable lotions or creams containing the active compound suspended or dissolved in, for example, a mixture of one or more of the following: mineral oil, sorbitan monostearate, polysorbate 60, cetyl ester wax, cetearyl alcohol, 2-octyldodecanol, benzyl alcohol and water.
- the compounds of the present invention may be administered by a single dose, multiple discrete doses or continuous infusion. Because the compounds preferably are small, easily diffusible and relatively stable, they can be well-suited to continuous infusion. Dose levels on the order of about 0.1 mg to about 10,000 mg of the active ingredient are useful in the treatment of the above conditions, with preferred levels being about 0.1 mg to about 1,000 mg.
- the specific dose level, and thus the therapeutically- effective amount, for any particular patient will vary depending upon a variety of factors, including the activity of the specific compound employed and its bioavailability at the site of drug action; the age, body weight, general health, sex and diet of the patient; the time of administration; the rate of excretion; drug combination; the severity of the particular disease being treated; and the form of administration.
- in vitro dosage-effect results provide useful guidance on the proper doses for patient administration. Studies in animal models also are helpful. The considerations for determining the proper dose levels are available to the skilled person. See Example 5 below.
- Certain compounds can administered in lyophilized form.
- 1 to 100 mg of a compound of the present invention may be lyophilized in individual vials, together with a carrier and a buffer, such as mannitol and sodium phospshate.
- the compound may be reconstituted in the vials with bacteriostatic water before administration.
- the compounds of the present invention are preferably administered orally, rectally, parenterally or topically at least 1 to 6 times daily, and may follow an initial bolus dose of higher concentration.
- Administration Regimen and Timing are preferably administered orally, rectally, parenterally or topically at least 1 to 6 times daily, and may follow an initial bolus dose of higher concentration.
- any administration regimen regulating the timing and sequence of drug delivery can be used and repeated as necessary to effect treatment.
- Such regimen may include pretreatment and/or co- administration with additional therapeutic agents.
- the compounds should be administered to the affected cells as soon as possible. In situations where nervous insult is anticipated, the compounds should be administered before the expected nervous insult.
- Such simations of increased likelihood of nervous insult include surgery (for example, carotid endarterectomy, cardiac, vascular, aortic, orthopedic); endovascular procedures such as arterial catherization (for example, carotid, vertebral, aortic, cardia, renal, spinal, Adamkiewicz); injections of embolic agents; coils or balloons for hemostasis; interruptions of vascularity for treatment of brain lesions; and predisposing medical conditions such as crescendo transient ischemic attacks, emboli and sequential strokes.
- any compound for the treatment of stroke should adequately cross the blood-brain barrier and obtain sufficiently therapeutic levels within the brain and cerebral spinal fluid.
- the compounds of the present invention may be administered (i) prior to surgery or radiation treatment to reduce the risk of metastasis; (ii) during surgery or in conjunction with radiation treatment; and/or (iii) after surgery or radiation therapy to reduce the risk of recurrence and to inhibit the growth of any residual tumorous cells.
- the compounds of the present invention may be administered as a continuous supplement to, or as a replacement for, homonal ablation in order to slow tumor cell growth in both the untreated primary tumor and the existing metastatic lesions.
- the compounds, methods and uses of the present invention are particularly useful where shed cells could not be removed by surgical intervention. After post-surgical recovery, the compounds, methods and uses of the present invention would be effective in reducing the chances of recurrence of a tumor engendered by such shed cells.
- the compounds, methods and uses of the present invention also to provide combined preparation for simultaneous, separate, or sequential use which contain other biologically active agents.
- biologically active agent can be either another compound of the present invention; steroids, for example hydrocortisomers such as methylprednisolone; anti- inflammatory or anti-immune drugs, such as methotrexate, azathioprine, cyclophosphamide or cyclosporin A; interferon- ⁇ ; antibodies, such as anti-CD4 antibodies; agents which can reduce the risk of a second ischemic event, such as ticlopidine; chemotherapeutic compositions; immunotherapeutic compositions; morphine for treating pain; or mixtures thereof.
- the compounds according to the invention include various substitutions available to the skilled person and are to be employed in accordance with the teachings contained herein.
- the Core Structures which constitute or include proline mimetics, can include a variety of functional groups as taught herein.
- the inventions include isosteres of the compounds or the function groups contained therein. Guiding principles and illustrative examples of functional groups and isosteres are set forth in Smith et al. , INTRODUCTION TO THE PRINCIPLES OF DRUG DESIGN (John Wright & Sons, Ltd.), which is hereby incorporated by reference.
- the compounds used according to the invention preferably are or contain moieties that resemble proline within their core strucmres. That is, these compounds are or contain proline mimetics.
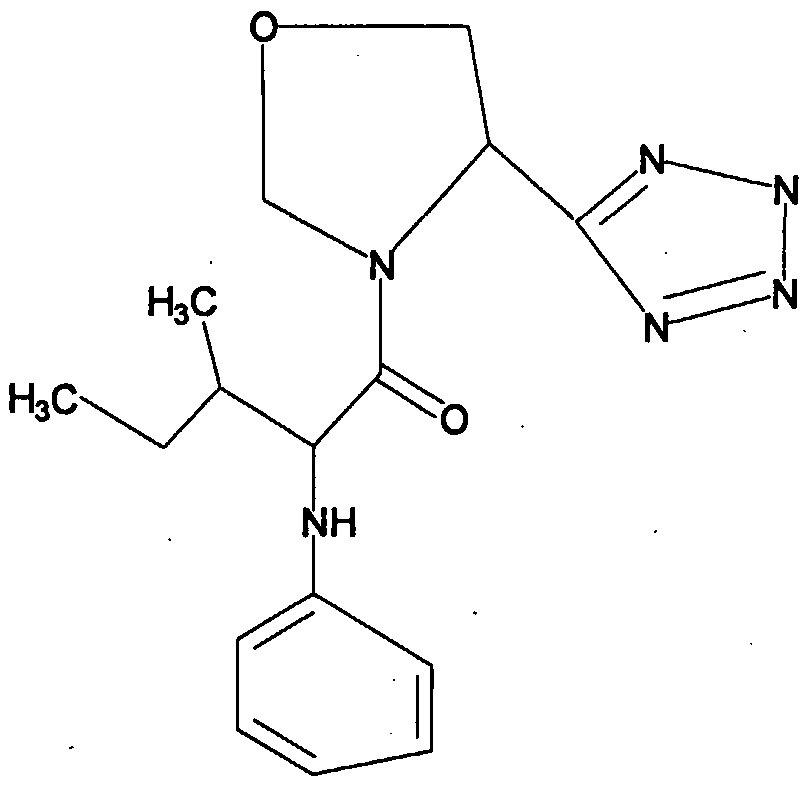
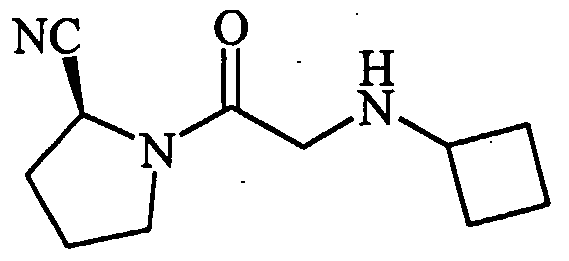
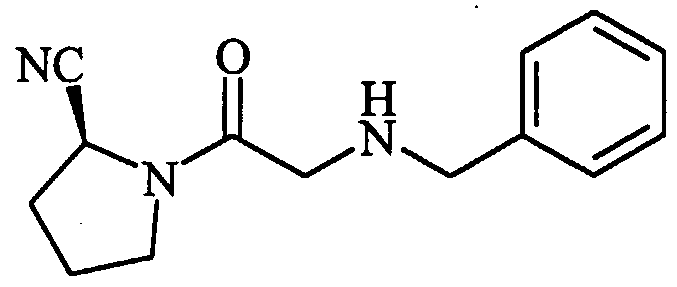
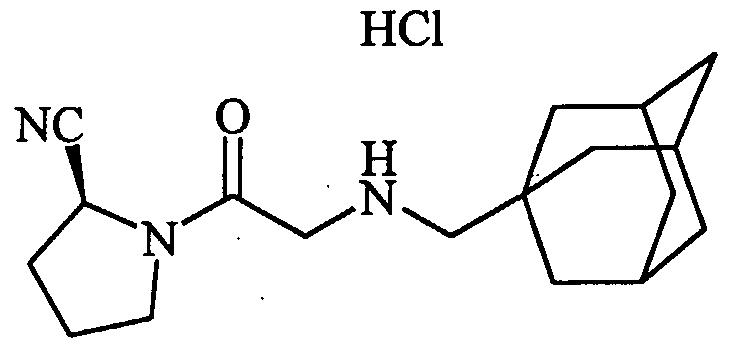
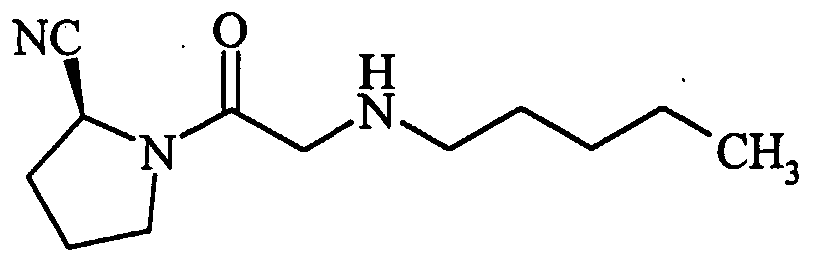
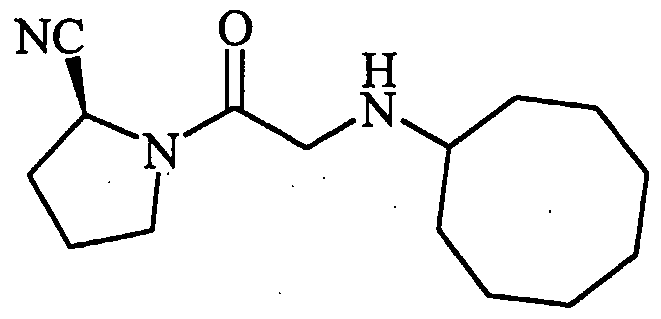
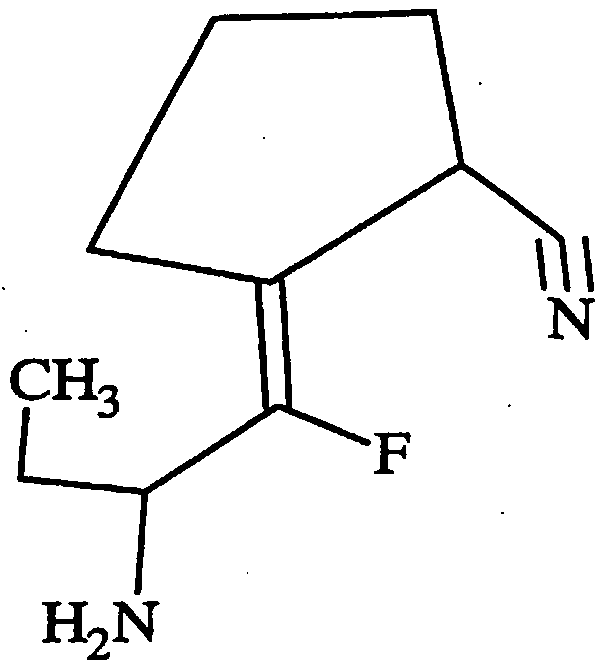
- One such compound that can be used according to the invention contains a proline mimetic and has the following structure:
- c-KPG This compound, referred to as "c-KPG,” was tested in an assay employing organotypic spinal motor neurons and threohydroxyaspartate (“THA”), which is an inhibitor of the glutamate reuptake receptor. Synthesis protocols for c-KPG are disclosed in Nguyen et al., J. Med. Chem. 41: 2100-10 (1998).
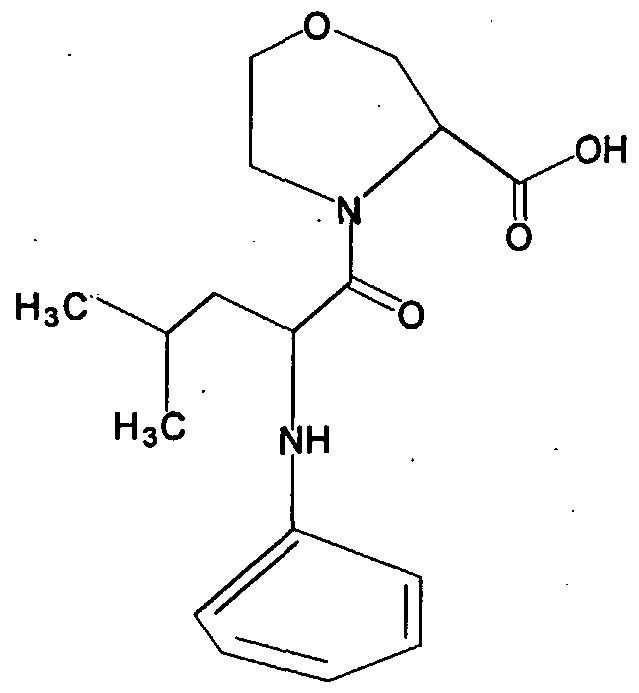
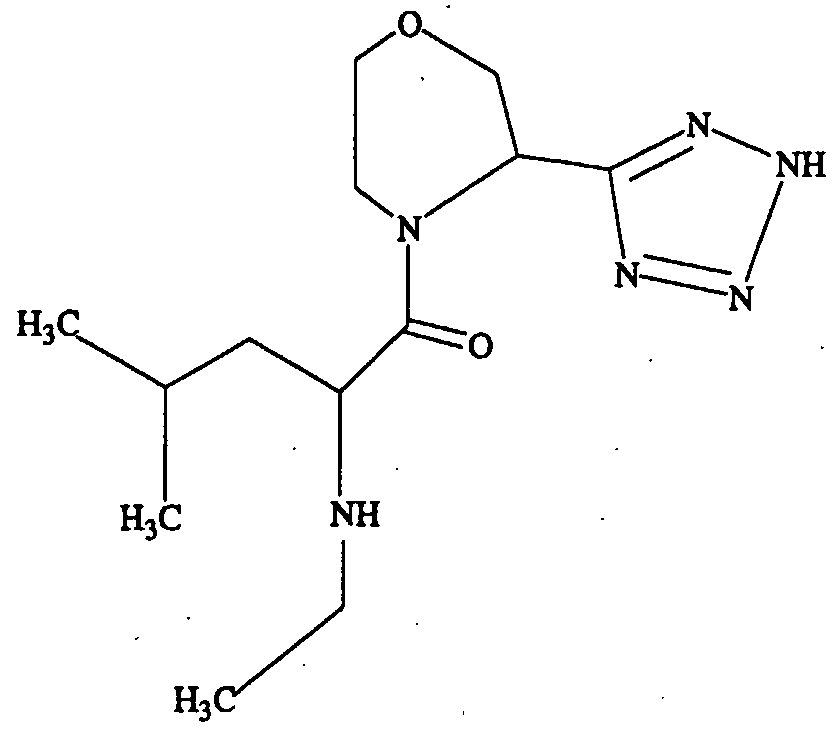
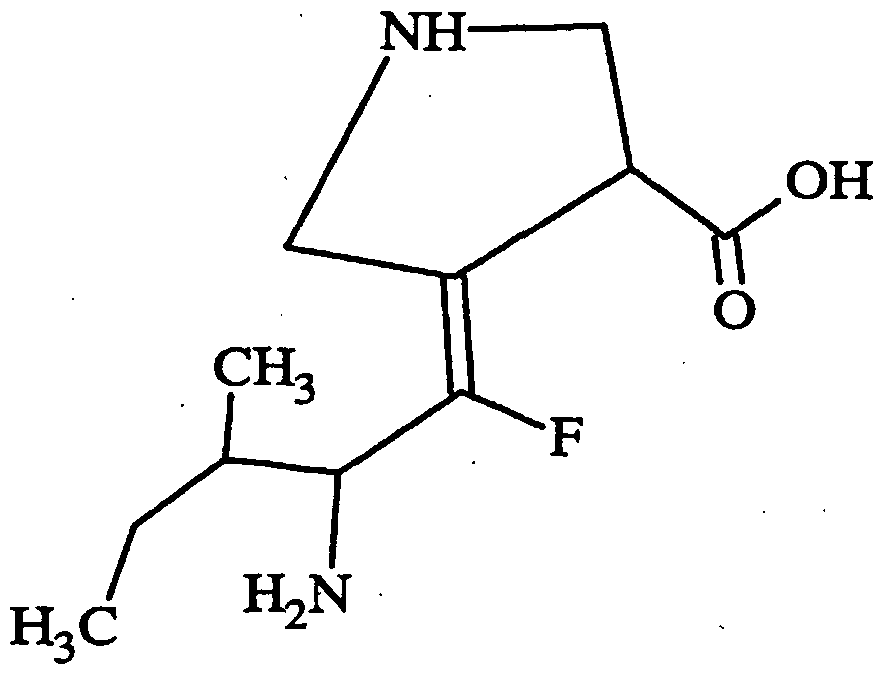
- Core structures which are DPP IV inhibitors and constitute or contain proline mimetics, are set forth below.
- Exemplary core structures are depicted schematically, and the functional/substitution groups are set forth in text. All substitutions contemplated herein are permissive for various provisos, either alone or in any combination, such that if one group is included in a given position another group at the same or different position can be excluded.
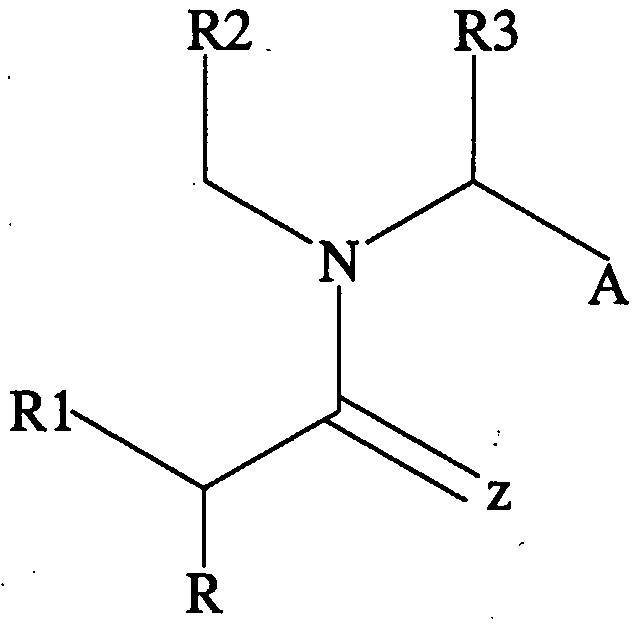
- Core Structure I is:
- X is CR2R3, O, S, or NR4; optional proviso 1 that if X is S, then A cannot be CN; optional proviso 2 that if X is CH 2 and R is H, then A cannot be C; optional proviso 3 that if X is S, then Rl cannot be amino-substimted alkyl; optional proviso 4 that if X is CH 2 , then A cannot be COOH; optional proviso 5 that if X is S, or if X and XI are both CH 2 , and Z is O, and A is CN, and Rl is H, then R is not NH substituted with C1-C9 straight or branched chain alkyl, or NH substituted with C3-C7 cycloalkyl;
- X is CR2R3, O, S, or NR4 with optional proviso 6 that X and XI cannot both be a heteroatom; optional proviso 7 if X and XI are both CH 2 , and Z is O, and Rl is NH 2 , then R is not 1-methylpropyl if A is COOH, and R is not cyclopentyl if A is CN.
- A is H, COOH, or isosteres of carboxylic acids, such as one selected from the group consisting of CN, SO 3 H, CONOH, PO 3 R5R6, SO 2 NHR7, tetrazole, amides, esters, and acid anhydrides with optional proviso 8 that if A is CN, and Rl is NH 2 , and Z is O, and R is 1-methylpropyl, then X and XI are not both CH 2 , X and XI are not S, X is not O, and Z is O or S;
- R and Rl are independently selected from the group of functional groups consisting of H, C r C 9 branched or straight chain alkyl, C 2 -C 9 branched or straight chain alkenyl, C 3 - C 8 cycloalkyl, C 5 -C 7 cycloalkenyl, aryl, heteroaryl and amino, wherein any of the functional groups can be substituted with one or more of C r C 9 straight or branched chain alkyl, aryl, heteroaryl, amino, halo, carbonyl, C r C 9 alkoxy, C 2 -C 9 alkenyloxy, phenoxy, benzyloxy, C 3 -C 8 cycloalkyl, cyano, amido, thiol, trifluromethyl, or hydroxy, wherein each of R and Rl can be the same or different; and
- R2, R3, R4, R5, R6 and R7, if present, are independently selected from the group of functional groups consisting of H, C r C 9 branched or straight chain alkyl, C 2 -C 9 branched or straight chain alkenyl, C 3 -C 8 cycloalkyl, C 5 -C 7 cycloalkenyl, aryl, heteroaryl and amino, wherein any of the functional groups can be substituted with one or more of - C 9 straight or branched chain alkyl, aryl, heteroaryl, amino, halo, carbonyl, C t -C 9 alkoxy, C 2 -C 9 alkenyloxy, phenoxy, benzyloxy, C 3 -C 8 cycloalkyl, cyano, amido, thiol, trifluromethyl, or hydroxy, wherein each of R2, R3, R4, R5, R6 and R7, if present, can be the same or different.
- core strucmres are provide according to the invention, such as those having ring modifications (II and HI): .
- X is CR2R3, O, S, or NR4;
- A is H, COOH, or isosteres of carboxylic acids, such as one selected from the group consisting of CN, SO 3 H, CONOH, PO 3 R5R6, SO 2 NHR7, tetrazole, amides, esters, and acid anhydrides;
- R and Rl are independently selected from the group of functional groups consisting of H, C,-C 9 branched or straight chain alkyl, C 2 -C 9 branched or straight chain alkenyl, C 3 - C 8 cycloalkyl, C 5 -C 7 cycloalkenyl, aryl, heteroaryl and amino, wherein any of the functional groups can be substituted with one or more of C r C 9 straight or branched chain alkyl, aryl, heteroaryl, amino, halo, carbonyl, C r C 9 alkoxy, C 2 -C 9 alkenyloxy, phenoxy, benzyloxy, C 3 -C 8 cycloalkyl, cyano, amido, thiol, trifluromethyl, or hydroxy, wherein each of R and Rl can be the same or different; and
- R2, R3, R4, R5, R6 and R7, if present, are independently selected from the group of functional groups consisting of H, C,-C 9 branched or straight chain alkyl, C 2 -C 9 branched or straight chain alkenyl, C 3 -C 8 cycloalkyl, C 5 -C 7 cycloalkenyl, aryl, heteroaryl and amino, wherein any of the functional groups can be substituted with one or more of C r C 9 straight or branched chain alkyl, aryl, heteroaryl, amino, halo, carbonyl, C,-C 9 alkoxy, C 2 -C 9 alkenyloxy, phenoxy, benzyloxy, C 3 -C 8 cycloalkyl, cyano, amido, thiol, trifluromethyl, or hydroxy, wherein each of R2, R3, R4, R5, R6 and R7, if present, can be the same or different.
- Core Structure in is:
- A is H, COOH, or isosteres of carboxylic acids, such as one selected from the group consisting of CN, SO 3 H, CONOH, PO 3 R5R6, SO 2 NHR7, tetrazole, amides, esters, and acid anhydrides;
- R, Rl, R2 and R3 are independently selected from the group of functional groups consisting of H, C r C 9 branched or straight chain alkyl, C 2 -C 9 branched or straight chain alkenyl, C 3 -C 8 cycloalkyl, C 5 -C 7 cycloalkenyl, aryl, heteroaryl and amino, wherein any of the functional groups can be substituted with one or more of C C 9 straight or branched chain alkyl, aryl, heteroaryl, amino, halo, carbonyl, C r C 9 alkoxy, C 2 -C 9 alkenyloxy, phenoxy, benzyloxy, C 3 -C 8 cycloalkyl, cyano, amido, thiol, trifluromethyl, or hydroxy, wherein each of R, Rl, R2 and R3 can be the same or different; and
- R4, R5, R6 and R7, if present, are independently selected from the group of functional groups consisting of H, C,-C 9 branched or straight chain alkyl, C 2 -C 9 branched or straight chain alkenyl, C 3 -C 8 cycloalkyl, C 5 -C 7 cycloalkenyl, aryl, heteroaryl and amino, wherein any of the functional groups can be substimted with one or more of C r C 9 straight or branched chain alkyl, aryl, heteroaryl, amino, halo, carbonyl, C r C 9 alkoxy, C 2 -C 9 alkenyloxy, phenoxy, benzyloxy, C 3 -C 8 cycloalkyl, cyano, amido, thiol, trifluromethyl, or hydroxy, wherein each of R4, R5, R6 and R7, if present, can be the same or different.
- X is CR2R3, O, S, or NR4;
- X is CR2R3, O, S, or NR4 with the proviso that X and XI cannot both be a heteroatom;
- A is H, COOH, or isosteres of carboxylic acids, such as one selected from the group consisting of CN, SO 3 H, CONOH, PO 3 R5R6, SO 2 NHR7, tetrazole, amides, esters, and acid anhydrides;
- R and Rl are independently selected from the group of functional groups consisting of H, C r C 9 branched or straight chain alkyl, C 2 -C 9 branched or straight chain alkenyl, C 3 - C 8 cycloalkyl, C 5 -C 7 cycloalkenyl, aryl, heteroaryl and amino, wherein any of the functional groups can be substituted with one or more of C r C 9 straight or branched chain alkyl, aryl, heteroaryl, amino, halo, carbonyl, C C 9 alkoxy, C 2 -C 9 alkenyloxy, phenoxy, benzyloxy, C 3 -C 8 cycloalkyl, cyano, amido, thiol, trifluromethyl, or hydroxy, wherein each of R and Rl can be the same or different; and
- R2, R3, R4, R5, R6 and R7, if present, are independently selected from the group of functional groups consisting of H, C r C 9 branched or straight chain alkyl, C 2 -C 9 branched or straight chain alkenyl, C 3 -C 8 cycloalkyl, C 5 -C 7 cycloalkenyl, aryl, heteroaryl and amino, wherein any of the functional groups can be substituted with one or more of C r C 9 straight or branched chain alkyl, aryl, heteroaryl, amino, halo, carbonyl, C r C 9 alkoxy, C 2 -C 9 alkenyloxy, phenoxy, benzyloxy, C 3 -C 8 cycloalkyl, cyano, amido, thiol, trifluromethyl, or hydroxy, wherein each of R2, R3, R4, R5, R6 and R7, if present, can be the same or different.
- the compounds of the core structures according to the present invention can administered in ester or salt forms according to the teachings provided herein. Acceptable formulations, dosages and administration regimens can be determined in accordance with the teachings contained herein.
- R is NH-R 1 ;
- R 1 is: C t - C 12 straight or branched chain alkyl; C 3 - C 7 cycloalkyl; CH 2 - CH 2 -NH-R U ;
- R" is a pyridine ring optionally substimted in one or two positions with halo, trifluoromethyl, cyano or nitro; or a pyrimidine ring optionally substimted in one position with halo, trifluromethyl, cyano or nitro;
- R ⁇ is a phenyl ring optionally substimted in one to three positions with halo or C, - C 3 alkoxy;
- Each R w is independently a phenyl ring optionally substimted in one position with halo or C, - C 3 alkoxy; and R v is a 2-oxopyrrolidine group or a C 2 - C 4 alkoxy group.
- R is NH-R 1 ;
- R 1 is: Cj - C 12 straight or branched chain alkyl optionally substimted with hydroxy, acetyl,
- adamantyl indanyl; piperidyl optionally substimted with benzyl; pyrrolidine optionally substimted with benzyl; bicycloheptyl optionally substituted in one to three positions with methyl; phenyl optionally substimted with in one to three positions with halo, methoxy, trifluoromethyl; pyridyl optionally substimted in one to three positions with halo, trifluoromethyl, nitro; or pyrimidyl optionally substimted with halo, trifluoromethyl, nitro;
- R m is phenyl optionally substituted with CN, or pyridyl optionally substimted with CN;
- R w is a group selected from phenyl, naphthyl, cyclohexenyl, pyridyl, pyrimidyl, adamantyl, phenoxy, wherein the group is optionally substimted in one to two positions with ethoxy, methoxy, halo, phenylsulfide, or phenylsulfide substimted with hydroxymethyl.
- Prlnclpal Group carboxylic acid
- Prlncloal Group carboxylic acid
- Parent Hvdride pyrrolidine
- EXAMPLE 5 EXEMPLARY NEURO ACTIVITY TESTING PROTOCOLS
- DRG dorsal root ganglion
- the spinal cord with attached DRGs from an adult mouse (15-10g) is removed.
- Spinal nerves are cut away using micro-dissection scissors and any excess material is trimmed until the DRG is free.
- Using sharp micro-dissecting scissors a transverse cut is made in the peripheral nerve, leaving 1-2 mm attached, and the explant is placed into Petri dish and covered with plating media.
- the spinal nerve is trimmed to about ImM in length. Then, embed the explant in 30 ⁇ L of reduced growth factor Matrigel on a circular coverslip, and place in a 35 mM culture dish.
- Primary antibody for example, Beta tubulin, Sigma Chemical Co.
- secondary antibody Alexa 488 Goat Anti-Mouse
- Cultures are derived from postnatal day 8 (P8) Sprague-Dawley rat lumbar spinal cord slices of 325 micron thickness. Each experiment consists of two 6- well plates with 5 slices from 4 different animals per well. Media changes are performed every 3 to 4 days. Cultures are treated with THA [L(-)-threo-3-hydroxyaspartic acid; Tocris Cookson Inc., Ballwin, Missouri] at 200 ⁇ M + compound (lO ⁇ M) after one week in culture.
- the control is an untreated sample with 0.1 % DMSO as vehicle.
- the THA control is a THA treated sample with 0.1 % DSMO as vehicle. Two wells are used per condition. One media change with new THA and compounds is performed.
- the experiment is stopped 6 to 8 days following drug treatment (13-15 total days in vitro, DIV) as dictated by visual assessment of lesion, by fixation with 4% paraformaldehyde/0.1 M phosphate buffer for 30 minutes.
- Slices are permeabilized with 100% cold methanol forlO minutes. Slices are transferred to staining wells. The slices are blocked with 10% HS/TBS. Primary antibody incubation is overnight at 4°C with SMI-32 antibody 1:5000 in 2% HS/TBS. SMI-32 was specific towards unphosphorylated H neurofilament subunit.
- Vectastain ABC Elite Kit with rat absorbed anti-mouse secondary antibody is used with DAB to stain the slices.
- the slices are mounted onto a slide and a coverslip is sealed with DPX mounting solution.
- Quantification of surviving neurons is performed on a Ziess Axiovert microscope. Neuronal survival is determined by observing an intact neuronal cell body with processes located ventrally of the central canal in each hemisphere. This correlates to laminae VII, VIII and IX. Each hemisphere is counted individually. The statistics can be performed with StatView software on a minimum of three different experiments per condition and significance should be determined as compared to THA control. The percent of protection can be determined from the average number of living neurons by the following equation:
- EXAMPLE 6 EXEMPLARY TESTING PROTOCOLS FOR PROSTATE
- Protocols for testing efficacy, dosing, and administration schedules for post- prostatectomy nerve recovery can be performed in accordance with the teachings of Example 5.
- DPP IV inhibitors in the treatment of prostate cancer, there are several cancer cell lines available of conducting in vitro assays. Appropriate cell lines include LNCaP, PC3, DU-145 and TSUPrl for use in cell proliferation assays.
- a cell line can be propagated in a standard medium, such as RPMI
Abstract
Description






























































































































Claims









Priority Applications (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| AU19164/01A AU1916401A (en) | 1999-11-12 | 2000-11-13 | Dipeptidyl peptidase iv inhibitors and methods of making and using dipeptidyl peptidase iv inhibitors |
| JP2001536541A JP2003535034A (en) | 1999-11-12 | 2000-11-13 | Dipeptidyl peptidase IV inhibitors and methods for producing and using dipeptidyl peptidase IV inhibitors |
| CA002390231A CA2390231A1 (en) | 1999-11-12 | 2000-11-13 | Dipeptidyl peptidase iv inhibitors and methods of making and using dipeptidyl peptidase iv inhibitors |
| EP00982093A EP1228061A4 (en) | 1999-11-12 | 2000-11-13 | Dipeptidyl peptidase iv inhibitors and methods of making and using dipeptidyl peptidase iv inhibitors |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US43908999A | 1999-11-12 | 1999-11-12 | |
| US09/439,089 | 1999-11-12 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2001034594A1 true WO2001034594A1 (en) | 2001-05-17 |
Family
ID=23743248
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2000/030836 WO2001034594A1 (en) | 1999-11-12 | 2000-11-13 | Dipeptidyl peptidase iv inhibitors and methods of making and using dipeptidyl peptidase iv inhibitors |
Country Status (5)
| Country | Link |
|---|---|
| EP (1) | EP1228061A4 (en) |
| JP (1) | JP2003535034A (en) |
| AU (1) | AU1916401A (en) |
| CA (1) | CA2390231A1 (en) |
| WO (1) | WO2001034594A1 (en) |
Cited By (110)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP1175213A2 (en) * | 1999-03-12 | 2002-01-30 | Bristol-Myers Squibb Company | Heterocyclic aromatic compounds useful as growth hormone secretagogues |
| WO2002083109A1 (en) * | 2001-04-11 | 2002-10-24 | Ferring Bv | Treatment of type 2 diabetes with inhibitors of dipeptidyl peptidase iv |
| WO2003002596A2 (en) * | 2001-06-27 | 2003-01-09 | Probiodrug Ag | Use of dipeptidyl peptidase iv inhibitors as therapeutics for neurological disorders |
| WO2003002553A2 (en) * | 2001-06-27 | 2003-01-09 | Smithkline Beecham Corporation | Fluoropyrrolidines as dipeptidyl peptidase inhibitors |
| WO2003015775A1 (en) * | 2001-08-17 | 2003-02-27 | Probiodrug Ag | New dipeptidyl peptidase iv inhibitors and their uses for lowering blood pressure levels |
| WO2003037327A1 (en) * | 2001-10-26 | 2003-05-08 | F. Hoffmann-La-Roche Ag | N-substituted pyrrolidin derivatives as dipeptidyl peptidase iv inhibitors |
| WO2003045228A2 (en) * | 2001-11-26 | 2003-06-05 | Trustees Of Tufts College | Methods for treating autoimmune disorders, and reagents related thereto |
| US6699871B2 (en) | 2001-07-06 | 2004-03-02 | Merck & Co., Inc. | Beta-amino heterocyclic dipeptidyl peptidase inhibitors for the treatment or prevention of diabetes |
| WO2003106456A3 (en) * | 2002-06-14 | 2004-03-04 | Sanofi Synthelabo | Azabicyclo-octane and nonane derivatives with ddp-iv inhibiting activity |
| US6710040B1 (en) | 2002-06-04 | 2004-03-23 | Pfizer Inc. | Fluorinated cyclic amides as dipeptidyl peptidase IV inhibitors |
| WO2004098591A2 (en) | 2003-05-05 | 2004-11-18 | Probiodrug Ag | Inhibitors of glutaminyl cyclase and their use in the treatment of neurological diseases |
| WO2004098625A2 (en) | 2003-05-05 | 2004-11-18 | Probiodrug Ag | Medical use of inhibitors of glutaminyl and glutamate cyclases |
| US6844316B2 (en) | 2001-09-06 | 2005-01-18 | Probiodrug Ag | Inhibitors of dipeptidyl peptidase I |
| WO2005012249A2 (en) | 2003-08-01 | 2005-02-10 | Bristol-Myers Squibb Company | Adamantyglycine- based inhibitors of dipeptidyl peptidase iv for the treatment of diabetes |
| EP1506776A1 (en) | 2003-08-12 | 2005-02-16 | KeyNeurotek AG | Use of enzyme inhibitors of the dipeptidylpeptidase IV and/or of the aminopeptidase n , and pharmaceutical preparations thereof for the prevention or therapy of neurodegenerative diseases |
| WO2005021536A2 (en) * | 2003-08-29 | 2005-03-10 | Sanofi-Aventis | Adamantane and azabicyclo-octane and nonane derivatives, process of their preparation and their use as dpp-iv inhibitors |
| US6890905B2 (en) | 2001-04-02 | 2005-05-10 | Prosidion Limited | Methods for improving islet signaling in diabetes mellitus and for its prevention |
| WO2005049027A2 (en) | 2003-11-03 | 2005-06-02 | Probiodrug Ag | Combinations useful for the treatment of neuronal disorders |
| WO2005079795A2 (en) * | 2004-02-20 | 2005-09-01 | Novartis Ag | Dpp-iv inhibitors for treating neurodegeneration and cognitive disorders |
| US6946480B2 (en) | 2002-02-28 | 2005-09-20 | Hans-Ulrich Demuth | Glutaminyl based DPIV inhibitors |
| US6949515B2 (en) | 1999-08-24 | 2005-09-27 | Probiodrug Ag | Effectors of dipeptidyl peptidase IV for topical use |
| US7078381B2 (en) | 1998-02-02 | 2006-07-18 | Trustees Of Tufts College | Method of regulating glucose metabolism, and reagents related thereto |
| US7098239B2 (en) | 2001-06-20 | 2006-08-29 | Merck & Co., Inc | Dipeptidyl peptidase inhibitors for the treatment of diabetes |
| US7101871B2 (en) | 2002-10-18 | 2006-09-05 | Merck & Co., Inc. | Beta-amino heterocyclic dipeptidyl peptidase inhibitors for the treatment or prevention of diabetes |
| US7109347B2 (en) | 2001-06-27 | 2006-09-19 | Probiodrug Ag | Dipeptidyl peptidase IV inhibitors and their uses as anti-cancer agents |
| US7122555B2 (en) | 2003-06-20 | 2006-10-17 | Hoffmann-La Roche Inc. | Pyrido [2,1-a] isoquinoline derivatives |
| US7132443B2 (en) | 2001-06-27 | 2006-11-07 | Smithklinebeecham Corporation | Fluoropyrrolidines as dipeptidyl peptidase inhibitors |
| US7157490B2 (en) | 2002-11-07 | 2007-01-02 | Merck & Co., Inc. | Phenylalanine derivatives as dipeptidyl peptidase inhibitors for the treatment or prevention of diabetes |
| US7166579B2 (en) | 1998-06-24 | 2007-01-23 | Prosidion Limited | Prodrugs of DP IV-inhibitors |
| US7169926B1 (en) | 2003-08-13 | 2007-01-30 | Takeda Pharmaceutical Company Limited | Dipeptidyl peptidase inhibitors |
| EP1760076A1 (en) * | 2005-09-02 | 2007-03-07 | Ferring B.V. | FAP Inhibitors |
| US7196201B2 (en) | 2001-06-27 | 2007-03-27 | Smithkline Beecham Corporation | Pyrrolidines as dipeptidyl peptidase inhibitors |
| US7205409B2 (en) | 2003-09-04 | 2007-04-17 | Abbott Laboratories | Pharmaceutical compositions as inhibitors of dipeptidyl peptidase-IV (DPP-IV) |
| US7208498B2 (en) | 2002-07-15 | 2007-04-24 | Merck & Co., Inc. | Piperidino pyrimidine dipeptidyl peptidase inhibitors for the treatment of diabetes |
| EA008226B1 (en) * | 2000-11-10 | 2007-04-27 | Тайсо Фармасьютикал Ко., Лтд. | Intermediates for the synthesis of cyanopyrrolidine derivatives |
| WO2007072083A1 (en) | 2005-12-23 | 2007-06-28 | Prosidion Limited | Treatment of type 2 diabetes with a combination of dpiv inhibitor and metformin or thiazolidinedione |
| US7238724B2 (en) | 2002-09-19 | 2007-07-03 | Abbott Laboratories | Pharmaceutical compositions as inhibitors of dipeptidyl peptidase-IV (DPP-IV) |
| US7253172B2 (en) | 2001-06-20 | 2007-08-07 | Merck & Co., Inc. | Dipeptidyl peptidase inhibitors for the treatment of diabetes |
| US7259160B2 (en) | 2003-07-31 | 2007-08-21 | Merck & Co., Inc. | Hexahydrodiazepinones as dipeptidyl peptidase-IV inhibitors for the treatment or prevention of diabetes |
| US7262207B2 (en) | 2002-09-19 | 2007-08-28 | Abbott Laboratories | Pharmaceutical compositions as inhibitors of dipeptidyl peptidase-IV (DPP-IV) |
| US7265128B2 (en) | 2003-01-17 | 2007-09-04 | Merck & Co., Inc. | 3-amino-4-phenylbutanoic acid derivatives as dipeptidyl peptidase inhibitors for the treatment or prevention of diabetes |
| WO2007120702A2 (en) | 2006-04-11 | 2007-10-25 | Arena Pharmaceuticals, Inc. | Use of gpr119 receptor agonists for increasing bone mass and for treating osteoporosis, and combination therapy relating thereto |
| US7304086B2 (en) | 2004-02-05 | 2007-12-04 | Probiodrug Ag | Inhibitors of glutaminyl cyclase |
| JPWO2005053695A1 (en) * | 2003-12-04 | 2007-12-06 | エーザイ・アール・アンド・ディー・マネジメント株式会社 | Agents for preventing or treating multiple sclerosis |
| KR100792275B1 (en) * | 2006-02-22 | 2008-01-08 | 영진약품공업주식회사 | Cyclic hydrazide derivatives having ?-amino group, its pharmaceutical acceptable salts and preparation process thereof |
| US7332520B2 (en) | 2003-06-06 | 2008-02-19 | Merck & Co., Inc. | Fused indoles as dipeptidyl peptidase inhibitors for the treatment or prevention of diabetes |
| US7368421B2 (en) | 2001-06-27 | 2008-05-06 | Probiodrug Ag | Use of dipeptidyl peptidase IV inhibitors in the treatment of multiple sclerosis |
| WO2008055945A1 (en) | 2006-11-09 | 2008-05-15 | Probiodrug Ag | 3-hydr0xy-1,5-dihydr0-pyrr0l-2-one derivatives as inhibitors of glutaminyl cyclase for the treatment of ulcer, cancer and other diseases |
| US7381537B2 (en) | 2003-05-05 | 2008-06-03 | Probiodrug Ag | Use of inhibitors of glutaminyl cyclases for treatment and prevention of disease |
| WO2008065141A1 (en) | 2006-11-30 | 2008-06-05 | Probiodrug Ag | Novel inhibitors of glutaminyl cyclase |
| US7388019B2 (en) | 2003-01-31 | 2008-06-17 | Merck & Co., Inc. | 3-amino-4-phenylbutanoic acid derivatives as dipeptidyl peptidase inhibitors for the treatment or prevention of diabetes |
| US7390809B2 (en) | 2002-10-07 | 2008-06-24 | Merck & Co., Inc. | Beta-amino heterocyclic dipeptidyl peptidase inhibitors for diabetes |
| US7456204B2 (en) | 2003-06-17 | 2008-11-25 | Merck & Co., Inc. | Cyclohexylglycine derivatives as dipeptidyl peptidase inhibitors for the treatment or prevention of diabetes |
| US7462599B2 (en) | 2003-10-15 | 2008-12-09 | Probiodrug Ag | Use of effectors of glutaminyl and glutamate cyclases |
| US7560455B2 (en) | 2003-05-14 | 2009-07-14 | Merck & Co., Inc. | 3-Amino-4-phenylbutanoic acid derivatives as dipeptidyl peptidase inhibitors for the treatment or prevention of diabetes |
| US7608577B2 (en) | 2001-10-12 | 2009-10-27 | Osi Pharmaceuticals, Inc. | Peptidyl ketones as inhibitors of DPIV |
| EP2116235A1 (en) | 2005-01-10 | 2009-11-11 | Arena Pharmaceuticals, Inc. | Combination therapy for the treatment of diabetes and conditions related thereto and for the treatment of conditions ameliorated by increasing a blood GLP-1 level |
| US7622496B2 (en) | 2005-12-23 | 2009-11-24 | Zealand Pharma A/S | Modified lysine-mimetic compounds |
| US7667044B2 (en) | 2003-11-03 | 2010-02-23 | Probiodrug Ag | Compounds for the treatment of neurological disorders |
| US7671073B2 (en) | 2004-05-18 | 2010-03-02 | Merck Sharp & Dohme Corp. | Cyclohexylalanine derivatives as dipeptidyl peptidase-IV inhibitors for the treatment or prevention of diabetes |
| US7678909B1 (en) | 2003-08-13 | 2010-03-16 | Takeda Pharmaceutical Company Limited | Dipeptidyl peptidase inhibitors |
| EP2165703A2 (en) | 2004-01-20 | 2010-03-24 | Novartis Pharma AG. | Direct compression formulation and process |
| US7687492B2 (en) | 2004-05-04 | 2010-03-30 | Merck Sharp & Dohme Corp. | 1,2,4-Oxadiazole derivatives as dipeptidyl peptidase-IV inhibitors for the treatment or prevention of diabetes |
| US7687625B2 (en) | 2003-03-25 | 2010-03-30 | Takeda Pharmaceutical Company Limited | Dipeptidyl peptidase inhibitors |
| US7687638B2 (en) | 2004-06-04 | 2010-03-30 | Takeda San Diego, Inc. | Dipeptidyl peptidase inhibitors |
| US7718666B2 (en) | 2003-06-20 | 2010-05-18 | Hoffmann-La Roche Inc. | Pyrido [2,1-a] isoquinoline derivatives |
| US7723344B2 (en) | 2003-08-13 | 2010-05-25 | Takeda San Diego, Inc. | Dipeptidyl peptidase inhibitors |
| US7727964B2 (en) | 2001-11-26 | 2010-06-01 | Trustees Of Tufts College | Peptidomimetic inhibitors of post-proline cleaving enzymes |
| US7728146B2 (en) | 2006-04-12 | 2010-06-01 | Probiodrug Ag | Enzyme inhibitors |
| US7732446B1 (en) | 2004-03-11 | 2010-06-08 | Takeda Pharmaceutical Company Limited | Dipeptidyl peptidase inhibitors |
| US7732162B2 (en) | 2003-05-05 | 2010-06-08 | Probiodrug Ag | Inhibitors of glutaminyl cyclase for treating neurodegenerative diseases |
| US7754757B2 (en) | 2004-02-05 | 2010-07-13 | Kyorin Pharmaceutical Co., Ltd. | Bicycloester derivative |
| US7781584B2 (en) | 2004-03-15 | 2010-08-24 | Takeda Pharmaceutical Company Limited | Dipeptidyl peptidase inhibitors |
| US7790734B2 (en) | 2003-09-08 | 2010-09-07 | Takeda Pharmaceutical Company Limited | Dipeptidyl peptidase inhibitors |
| US7825242B2 (en) | 2004-07-16 | 2010-11-02 | Takeda Pharmaceutical Company Limted | Dipeptidyl peptidase inhibitors |
| WO2011005929A1 (en) | 2009-07-09 | 2011-01-13 | Arena Pharmaceuticals, Inc. | Piperidine derivative and its use for the treatment of diabets and obesity |
| US7872124B2 (en) | 2004-12-21 | 2011-01-18 | Takeda Pharmaceutical Company Limited | Dipeptidyl peptidase inhibitors |
| WO2011029920A1 (en) | 2009-09-11 | 2011-03-17 | Probiodrug Ag | Heterocylcic derivatives as inhibitors of glutaminyl cyclase |
| US7915427B2 (en) | 2006-03-08 | 2011-03-29 | Kyorin Pharmaceuticals Co., Ltd. | Process for producing aminoacetyl pyrrolidine carbonitrile derivative and intermediate for production thereof |
| US7960384B2 (en) | 2006-03-28 | 2011-06-14 | Takeda Pharmaceutical Company Limited | Dipeptidyl peptidase inhibitors |
| WO2011107530A2 (en) | 2010-03-03 | 2011-09-09 | Probiodrug Ag | Novel inhibitors |
| WO2011110613A1 (en) | 2010-03-10 | 2011-09-15 | Probiodrug Ag | Heterocyclic inhibitors of glutaminyl cyclase (qc, ec 2.3.2.5) |
| WO2011127051A1 (en) | 2010-04-06 | 2011-10-13 | Arena Pharmaceuticals, Inc. | Modulators of the gpr119 receptor and the treatment of disorders related thereto |
| WO2011131748A2 (en) | 2010-04-21 | 2011-10-27 | Probiodrug Ag | Novel inhibitors |
| US8084605B2 (en) | 2006-11-29 | 2011-12-27 | Kelly Ron C | Polymorphs of succinate salt of 2-[6-(3-amino-piperidin-1-yl)-3-methyl-2,4-dioxo-3,4-dihydro-2H-pyrimidin-1-ylmethy]-4-fluor-benzonitrile and methods of use therefor |
| US8093236B2 (en) | 2007-03-13 | 2012-01-10 | Takeda Pharmaceuticals Company Limited | Weekly administration of dipeptidyl peptidase inhibitors |
| US8143427B2 (en) | 2007-03-22 | 2012-03-27 | Kyorin Pharmaceutical Co., Ltd. | Method for producing aminoacetylpyrrolidinecarbonitrile derivative |
| WO2012040279A1 (en) | 2010-09-22 | 2012-03-29 | Arena Pharmaceuticals, Inc. | Modulators of the gpr119 receptor and the treatment of disorders related thereto |
| US8222411B2 (en) | 2005-09-16 | 2012-07-17 | Takeda Pharmaceutical Company Limited | Dipeptidyl peptidase inhibitors |
| WO2012123563A1 (en) | 2011-03-16 | 2012-09-20 | Probiodrug Ag | Benz imidazole derivatives as inhibitors of glutaminyl cyclase |
| WO2012135570A1 (en) | 2011-04-01 | 2012-10-04 | Arena Pharmaceuticals, Inc. | Modulators of the gpr119 receptor and the treatment of disorders related thereto |
| WO2012145604A1 (en) | 2011-04-22 | 2012-10-26 | Arena Pharmaceuticals, Inc. | Modulators of the gpr119 receptor and the treatment of disorders related thereto |
| WO2012145361A1 (en) | 2011-04-19 | 2012-10-26 | Arena Pharmaceuticals, Inc. | Modulators of the gpr119 receptor and the treatment of disorders related thereto |
| WO2012145603A1 (en) | 2011-04-22 | 2012-10-26 | Arena Pharmaceuticals, Inc. | Modulators of the gpr119 receptor and the treatment of disorders related thereto |
| US8324383B2 (en) | 2006-09-13 | 2012-12-04 | Takeda Pharmaceutical Company Limited | Methods of making polymorphs of benzoate salt of 2-[[6-[(3R)-3-amino-1-piperidinyl]-3,4-dihydro-3-methyl-2,4-dioxo-1(2H)-pyrimidinyl]methyl]-benzonitrile |
| WO2012170702A1 (en) | 2011-06-08 | 2012-12-13 | Arena Pharmaceuticals, Inc. | Modulators of the gpr119 receptor and the treatment of disorders related thereto |
| WO2013055910A1 (en) | 2011-10-12 | 2013-04-18 | Arena Pharmaceuticals, Inc. | Modulators of the gpr119 receptor and the treatment of disorders related thereto |
| WO2014074668A1 (en) | 2012-11-08 | 2014-05-15 | Arena Pharmaceuticals, Inc. | Modulators of gpr119 and the treatment of disorders related thereto |
| WO2014113750A1 (en) | 2013-01-18 | 2014-07-24 | Dnj Pharma, Inc. | Novel ddp-iv inhibitors |
| US8883714B2 (en) | 2008-04-07 | 2014-11-11 | Arena Pharmaceuticals, Inc. | Pharmaceutical compositions comprising GPR119 agonists which act as peptide YY (PYY) secretagogues |
| US8906901B2 (en) | 2005-09-14 | 2014-12-09 | Takeda Pharmaceutical Company Limited | Administration of dipeptidyl peptidase inhibitors |
| US8927590B2 (en) | 2006-12-21 | 2015-01-06 | Zealand Pharma A/S | Synthesis of pyrrolidine compounds |
| EP2839832A2 (en) | 2003-11-17 | 2015-02-25 | Novartis AG | Use of dipeptidyl peptidase IV inhibitors |
| EP2865670A1 (en) | 2007-04-18 | 2015-04-29 | Probiodrug AG | Thiourea derivatives as glutaminyl cyclase inhibitors |
| WO2018162722A1 (en) | 2017-03-09 | 2018-09-13 | Deutsches Institut Für Ernährungsforschung Potsdam-Rehbrücke | Dpp-4 inhibitors for use in treating bone fractures |
| EP3461819A1 (en) | 2017-09-29 | 2019-04-03 | Probiodrug AG | Inhibitors of glutaminyl cyclase |
| US10555929B2 (en) | 2015-03-09 | 2020-02-11 | Coherus Biosciences, Inc. | Methods for the treatment of nonalcoholic fatty liver disease and/or lipodystrophy |
| US11253508B2 (en) | 2017-04-03 | 2022-02-22 | Coherus Biosciences, Inc. | PPARy agonist for treatment of progressive supranuclear palsy |
| US11324799B2 (en) | 2017-05-05 | 2022-05-10 | Zealand Pharma A/S | Gap junction intercellular communication modulators and their use for the treatment of diabetic eye disease |
| EP4000630A1 (en) | 2014-09-03 | 2022-05-25 | Invex Therapeutics Ltd | Elevated intracranial pressure treatment |
Families Citing this family (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| TW583185B (en) * | 2000-06-13 | 2004-04-11 | Novartis Ag | N-(substituted glycyl)-2-cyanopyrrolidines and pharmaceutical composition for inhibiting dipeptidyl peptidase-IV (DPP-IV) or for the prevention or treatment of diseases or conditions associated with elevated levels of DPP-IV comprising the same |
| RU2396269C2 (en) * | 2005-12-01 | 2010-08-10 | Ф.Хоффманн-Ля Рош Аг | Heteroaryl-substituted piperidine derivatives as hepatic carnitine palmitoyltransferase (l-cpt1) inhibitors |
| EP1971862B1 (en) | 2006-04-11 | 2010-11-10 | Arena Pharmaceuticals, Inc. | Methods of using gpr119 receptor to identify compounds useful for increasing bone mass in an individual |
| KR100848491B1 (en) * | 2007-01-16 | 2008-07-28 | 영진약품공업주식회사 | 2-thiazolidine derivatives having beta;-amino group, pharmaceutical acceptable salts and preparation process thereof |
| CA2732984A1 (en) | 2008-08-07 | 2010-02-11 | Kyorin Pharmaceutical Co., Ltd. | Process for production of bicyclo[2.2.2]octylamine derivative |
Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4684662A (en) * | 1983-01-07 | 1987-08-04 | Hoechst Aktiengesellschaft | Disubstituted proline derivatives |
| US4691022A (en) * | 1983-06-23 | 1987-09-01 | Hoechst Aktiengesellschaft | Process for the preparation of monocyclic, bicyclic and tricyclic aminoacids |
| US4743616A (en) * | 1984-07-31 | 1988-05-10 | Suntory Limited | Novel biologically active compound having anti-prolyl endopeptidase activity |
| US5118811A (en) * | 1989-04-13 | 1992-06-02 | Japan Tobacco Inc. | Amino acid derivatives possessing prolyl endopeptidase-inhibitory activities |
| WO1998019998A2 (en) * | 1996-11-07 | 1998-05-14 | Novartis Ag | N-substituted 2-cyanopyrrolidines |
Family Cites Families (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DD296075A5 (en) * | 1989-08-07 | 1991-11-21 | Martin-Luther-Universitaet Halle-Wittenberg,De | PROCESS FOR THE PREPARATION OF NEW INHIBITORS OF DIPEPTIDYL PEPTIDASE IV |
| CA2121369C (en) * | 1991-10-22 | 2003-04-29 | William W. Bachovchin | Inhibitors of dipeptidyl-aminopeptidase type iv |
| IL111785A0 (en) * | 1993-12-03 | 1995-01-24 | Ferring Bv | Dp-iv inhibitors and pharmaceutical compositions containing them |
| US5543396A (en) * | 1994-04-28 | 1996-08-06 | Georgia Tech Research Corp. | Proline phosphonate derivatives |
| WO1995034538A2 (en) * | 1994-06-10 | 1995-12-21 | Universitaire Instelling Antwerpen | Purification of serine proteases and synthetic inhibitors thereof |
| DE19616486C5 (en) * | 1996-04-25 | 2016-06-30 | Royalty Pharma Collection Trust | Method for lowering the blood glucose level in mammals |
| US6011155A (en) * | 1996-11-07 | 2000-01-04 | Novartis Ag | N-(substituted glycyl)-2-cyanopyrrolidines, pharmaceutical compositions containing them and their use in inhibiting dipeptidyl peptidase-IV |
| DE19828114A1 (en) * | 1998-06-24 | 2000-01-27 | Probiodrug Ges Fuer Arzneim | Produgs of unstable inhibitors of dipeptidyl peptidase IV |
| DE19828113A1 (en) * | 1998-06-24 | 2000-01-05 | Probiodrug Ges Fuer Arzneim | Prodrugs of Dipeptidyl Peptidase IV Inhibitors |
| CO5150173A1 (en) * | 1998-12-10 | 2002-04-29 | Novartis Ag | COMPOUNDS N- (REPLACED GLYCLE) -2-DIPEPTIDYL-IV PEPTIDASE INHIBITING CYANOPIRROLIDINS (DPP-IV) WHICH ARE EFFECTIVE IN THE TREATMENT OF CONDITIONS MEDIATED BY DPP-IV INHIBITION |
| US6110949A (en) * | 1999-06-24 | 2000-08-29 | Novartis Ag | N-(substituted glycyl)-4-cyanothiazolidines, pharmaceutical compositions containing them and their use in inhibiting dipeptidyl peptidase-IV |
-
2000
- 2000-11-13 JP JP2001536541A patent/JP2003535034A/en active Pending
- 2000-11-13 AU AU19164/01A patent/AU1916401A/en not_active Abandoned
- 2000-11-13 CA CA002390231A patent/CA2390231A1/en not_active Abandoned
- 2000-11-13 EP EP00982093A patent/EP1228061A4/en not_active Withdrawn
- 2000-11-13 WO PCT/US2000/030836 patent/WO2001034594A1/en active Search and Examination
Patent Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4684662A (en) * | 1983-01-07 | 1987-08-04 | Hoechst Aktiengesellschaft | Disubstituted proline derivatives |
| US4691022A (en) * | 1983-06-23 | 1987-09-01 | Hoechst Aktiengesellschaft | Process for the preparation of monocyclic, bicyclic and tricyclic aminoacids |
| US4743616A (en) * | 1984-07-31 | 1988-05-10 | Suntory Limited | Novel biologically active compound having anti-prolyl endopeptidase activity |
| US5118811A (en) * | 1989-04-13 | 1992-06-02 | Japan Tobacco Inc. | Amino acid derivatives possessing prolyl endopeptidase-inhibitory activities |
| WO1998019998A2 (en) * | 1996-11-07 | 1998-05-14 | Novartis Ag | N-substituted 2-cyanopyrrolidines |
Non-Patent Citations (1)
| Title |
|---|
| See also references of EP1228061A4 * |
Cited By (182)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8513190B2 (en) | 1998-02-02 | 2013-08-20 | Trustees Of Tufts College | Method of regulating glucose metabolism, and reagents related thereto |
| US7078381B2 (en) | 1998-02-02 | 2006-07-18 | Trustees Of Tufts College | Method of regulating glucose metabolism, and reagents related thereto |
| US9044424B2 (en) | 1998-02-02 | 2015-06-02 | 1149336 Ontario, Inc. | Methods of regulating glucose metabolism, and reagents related thereto |
| US7459428B2 (en) | 1998-02-02 | 2008-12-02 | Trustees Of Tufts College | Method of regulating glucose metabolism, and reagents related thereto |
| US8318669B2 (en) | 1998-02-02 | 2012-11-27 | Trustees Of Tufts College | Method of regulating glucose metabolism, and reagents related thereto |
| US7829530B2 (en) | 1998-02-02 | 2010-11-09 | Trustees Of Tufts College | Method of regulating glucose metabolism, and reagents related thereto |
| US7166579B2 (en) | 1998-06-24 | 2007-01-23 | Prosidion Limited | Prodrugs of DP IV-inhibitors |
| EP1175213A2 (en) * | 1999-03-12 | 2002-01-30 | Bristol-Myers Squibb Company | Heterocyclic aromatic compounds useful as growth hormone secretagogues |
| EP1175213A4 (en) * | 1999-03-12 | 2007-04-11 | Bristol Myers Squibb Co | Heterocyclic aromatic compounds useful as growth hormone secretagogues |
| US6949515B2 (en) | 1999-08-24 | 2005-09-27 | Probiodrug Ag | Effectors of dipeptidyl peptidase IV for topical use |
| US7335645B2 (en) | 1999-08-24 | 2008-02-26 | Probiodrug Ag | Effectors of dipeptidyl peptidase IV for topical use |
| EA008226B1 (en) * | 2000-11-10 | 2007-04-27 | Тайсо Фармасьютикал Ко., Лтд. | Intermediates for the synthesis of cyanopyrrolidine derivatives |
| US6890905B2 (en) | 2001-04-02 | 2005-05-10 | Prosidion Limited | Methods for improving islet signaling in diabetes mellitus and for its prevention |
| WO2002083109A1 (en) * | 2001-04-11 | 2002-10-24 | Ferring Bv | Treatment of type 2 diabetes with inhibitors of dipeptidyl peptidase iv |
| US7253172B2 (en) | 2001-06-20 | 2007-08-07 | Merck & Co., Inc. | Dipeptidyl peptidase inhibitors for the treatment of diabetes |
| US7098239B2 (en) | 2001-06-20 | 2006-08-29 | Merck & Co., Inc | Dipeptidyl peptidase inhibitors for the treatment of diabetes |
| WO2003002553A3 (en) * | 2001-06-27 | 2003-03-27 | Smithkline Beecham Corp | Fluoropyrrolidines as dipeptidyl peptidase inhibitors |
| US7132443B2 (en) | 2001-06-27 | 2006-11-07 | Smithklinebeecham Corporation | Fluoropyrrolidines as dipeptidyl peptidase inhibitors |
| CN1321682C (en) * | 2001-06-27 | 2007-06-20 | 前体生物药物股份有限公司 | New dipeptidyl peptidase iv inhibitors and their uses as anti-cancer agents |
| US7109347B2 (en) | 2001-06-27 | 2006-09-19 | Probiodrug Ag | Dipeptidyl peptidase IV inhibitors and their uses as anti-cancer agents |
| WO2003002595A2 (en) * | 2001-06-27 | 2003-01-09 | Probiodrug Ag | Dipeptidyl peptidase iv inhibitors and their uses as anti-cancer agents |
| WO2003002553A2 (en) * | 2001-06-27 | 2003-01-09 | Smithkline Beecham Corporation | Fluoropyrrolidines as dipeptidyl peptidase inhibitors |
| US7368421B2 (en) | 2001-06-27 | 2008-05-06 | Probiodrug Ag | Use of dipeptidyl peptidase IV inhibitors in the treatment of multiple sclerosis |
| US7183290B2 (en) | 2001-06-27 | 2007-02-27 | Smithkline Beecham Corporation | Fluoropyrrolidines as dipeptidyl peptidase inhibitors |
| WO2003002595A3 (en) * | 2001-06-27 | 2003-05-08 | Probiodrug Ag | Dipeptidyl peptidase iv inhibitors and their uses as anti-cancer agents |
| US7368576B2 (en) | 2001-06-27 | 2008-05-06 | Probiodrug Ag | Dipeptidyl peptidase IV inhibitors and their uses as anti-cancer agents |
| WO2003002596A2 (en) * | 2001-06-27 | 2003-01-09 | Probiodrug Ag | Use of dipeptidyl peptidase iv inhibitors as therapeutics for neurological disorders |
| US7196201B2 (en) | 2001-06-27 | 2007-03-27 | Smithkline Beecham Corporation | Pyrrolidines as dipeptidyl peptidase inhibitors |
| WO2003002596A3 (en) * | 2001-06-27 | 2003-07-31 | Probiodrug Ag | Use of dipeptidyl peptidase iv inhibitors as therapeutics for neurological disorders |
| US7125873B2 (en) | 2001-07-06 | 2006-10-24 | Merck & Co., Inc. | Beta-amino tetrahydroimidazo (1, 2-a) pyrazines and tetrahydrotrioazolo (4, 3-a) pyrazines as dipeptidyl peptidase inhibitors for the treatment or prevention of diabetes |
| US8440668B2 (en) | 2001-07-06 | 2013-05-14 | Merck Sharp & Dohme Corp | Beta-amino heterocyclic dipeptidyl peptidase inhibitors for the treatment of type 2 diabetes |
| US6699871B2 (en) | 2001-07-06 | 2004-03-02 | Merck & Co., Inc. | Beta-amino heterocyclic dipeptidyl peptidase inhibitors for the treatment or prevention of diabetes |
| US8168637B2 (en) | 2001-07-06 | 2012-05-01 | Merck Sharp & Dohme Corp. | Beta-amino heterocyclic dipeptidyl peptidase inhibitors for the treatment of diabetes |
| WO2003015775A1 (en) * | 2001-08-17 | 2003-02-27 | Probiodrug Ag | New dipeptidyl peptidase iv inhibitors and their uses for lowering blood pressure levels |
| US6844316B2 (en) | 2001-09-06 | 2005-01-18 | Probiodrug Ag | Inhibitors of dipeptidyl peptidase I |
| US7144856B2 (en) | 2001-09-06 | 2006-12-05 | Probiodrug Ag | Inhibitors of dipeptidyl peptidase I |
| US7608577B2 (en) | 2001-10-12 | 2009-10-27 | Osi Pharmaceuticals, Inc. | Peptidyl ketones as inhibitors of DPIV |
| WO2003037327A1 (en) * | 2001-10-26 | 2003-05-08 | F. Hoffmann-La-Roche Ag | N-substituted pyrrolidin derivatives as dipeptidyl peptidase iv inhibitors |
| US6861440B2 (en) | 2001-10-26 | 2005-03-01 | Hoffmann-La Roche Inc. | DPP IV inhibitors |
| US7314884B2 (en) | 2001-10-26 | 2008-01-01 | Hoffmann-La Roche Inc. | DPP IV inhibitors |
| US7803819B2 (en) | 2001-10-26 | 2010-09-28 | Hoffmann-La Roche Inc. | DPP IV inhibitors |
| EA007968B1 (en) * | 2001-10-26 | 2007-02-27 | Ф. Хоффманн-Ля Рош Аг | N-substituted pyrrolidin derivatives as dipeptidyl peptidase iv inhibitors |
| US8410053B2 (en) | 2001-11-26 | 2013-04-02 | Trustees Of Tufts College | Methods for treating autoimmune disorders, and reagents related thereto |
| WO2003045228A3 (en) * | 2001-11-26 | 2004-10-21 | Tufts College | Methods for treating autoimmune disorders, and reagents related thereto |
| US7727964B2 (en) | 2001-11-26 | 2010-06-01 | Trustees Of Tufts College | Peptidomimetic inhibitors of post-proline cleaving enzymes |
| US9757400B2 (en) | 2001-11-26 | 2017-09-12 | Trustees Of Tufts College | Peptidomimetic inhibitors of post-proline cleaving enzymes |
| WO2003045228A2 (en) * | 2001-11-26 | 2003-06-05 | Trustees Of Tufts College | Methods for treating autoimmune disorders, and reagents related thereto |
| EP2769715A3 (en) * | 2001-11-26 | 2014-09-17 | Trustees Of Tufts College | Methods for treating autoimmune disorders, and reagents related thereto |
| US6946480B2 (en) | 2002-02-28 | 2005-09-20 | Hans-Ulrich Demuth | Glutaminyl based DPIV inhibitors |
| US6710040B1 (en) | 2002-06-04 | 2004-03-23 | Pfizer Inc. | Fluorinated cyclic amides as dipeptidyl peptidase IV inhibitors |
| EA008166B1 (en) * | 2002-06-14 | 2007-04-27 | Санофи-Авентис | Azabicyclo-octane and nonane derivatives with ddp-iv inhibiting activity |
| CN100347170C (en) * | 2002-06-14 | 2007-11-07 | 赛诺菲-安万特公司 | Azabicyclo-octane and nonane derivatives with DPP-IV inhibiting activity |
| WO2003106456A3 (en) * | 2002-06-14 | 2004-03-04 | Sanofi Synthelabo | Azabicyclo-octane and nonane derivatives with ddp-iv inhibiting activity |
| US7781455B2 (en) | 2002-06-14 | 2010-08-24 | Sanofi-Aventis | Compounds |
| JP2005532369A (en) * | 2002-06-14 | 2005-10-27 | サノフイ−アベンテイス | Azabicyclo-octane and nonane derivatives having DDP-IV inhibitory activity |
| US7208498B2 (en) | 2002-07-15 | 2007-04-24 | Merck & Co., Inc. | Piperidino pyrimidine dipeptidyl peptidase inhibitors for the treatment of diabetes |
| US7238724B2 (en) | 2002-09-19 | 2007-07-03 | Abbott Laboratories | Pharmaceutical compositions as inhibitors of dipeptidyl peptidase-IV (DPP-IV) |
| US8637546B2 (en) | 2002-09-19 | 2014-01-28 | Abbvie Inc. | Pharmaceutical compositions as inhibitors of dipeptidyl peptidase-IV (DDP-IV) |
| US7262207B2 (en) | 2002-09-19 | 2007-08-28 | Abbott Laboratories | Pharmaceutical compositions as inhibitors of dipeptidyl peptidase-IV (DPP-IV) |
| US8119664B2 (en) | 2002-09-19 | 2012-02-21 | Abbott Laboratories | Pharmaceutical compositions as inhibitors of dipeptidyl peptidase-IV (DPP-IV) |
| US7390809B2 (en) | 2002-10-07 | 2008-06-24 | Merck & Co., Inc. | Beta-amino heterocyclic dipeptidyl peptidase inhibitors for diabetes |
| US7101871B2 (en) | 2002-10-18 | 2006-09-05 | Merck & Co., Inc. | Beta-amino heterocyclic dipeptidyl peptidase inhibitors for the treatment or prevention of diabetes |
| US7157490B2 (en) | 2002-11-07 | 2007-01-02 | Merck & Co., Inc. | Phenylalanine derivatives as dipeptidyl peptidase inhibitors for the treatment or prevention of diabetes |
| US7265128B2 (en) | 2003-01-17 | 2007-09-04 | Merck & Co., Inc. | 3-amino-4-phenylbutanoic acid derivatives as dipeptidyl peptidase inhibitors for the treatment or prevention of diabetes |
| US7388019B2 (en) | 2003-01-31 | 2008-06-17 | Merck & Co., Inc. | 3-amino-4-phenylbutanoic acid derivatives as dipeptidyl peptidase inhibitors for the treatment or prevention of diabetes |
| US7687625B2 (en) | 2003-03-25 | 2010-03-30 | Takeda Pharmaceutical Company Limited | Dipeptidyl peptidase inhibitors |
| EA009291B1 (en) * | 2003-05-05 | 2007-12-28 | Пробиодруг Аг | Use of effectors of glutaminyl and glutamate cyclases |
| AU2004237408C9 (en) * | 2003-05-05 | 2010-04-15 | Probiodrug Ag | Medical use of inhibitors of glutaminyl and glutamate cyclases |
| WO2004098591A2 (en) | 2003-05-05 | 2004-11-18 | Probiodrug Ag | Inhibitors of glutaminyl cyclase and their use in the treatment of neurological diseases |
| US7732162B2 (en) | 2003-05-05 | 2010-06-08 | Probiodrug Ag | Inhibitors of glutaminyl cyclase for treating neurodegenerative diseases |
| EP2206496A1 (en) | 2003-05-05 | 2010-07-14 | Probiodrug AG | Medical use of inhibitors of glutaminyl and glutamate cyclases |
| WO2004098625A3 (en) * | 2003-05-05 | 2005-06-23 | Probiodrug Ag | Medical use of inhibitors of glutaminyl and glutamate cyclases |
| AU2004237408C1 (en) * | 2003-05-05 | 2010-04-01 | Probiodrug Ag | Medical use of inhibitors of glutaminyl and glutamate cyclases |
| DE202004021723U1 (en) | 2003-05-05 | 2010-07-15 | Probiodrug Ag | Medical use of inhibitors of glutaminyl and glutamate cyclases |
| US7381537B2 (en) | 2003-05-05 | 2008-06-03 | Probiodrug Ag | Use of inhibitors of glutaminyl cyclases for treatment and prevention of disease |
| US8685664B2 (en) | 2003-05-05 | 2014-04-01 | Probiodrug Ag | Use of inhibtors of glutaminyl cyclases for treatment and prevention of disease |
| AU2004237408B2 (en) * | 2003-05-05 | 2009-10-01 | Probiodrug Ag | Medical use of inhibitors of glutaminyl and glutamate cyclases |
| US8809010B2 (en) | 2003-05-05 | 2014-08-19 | Probiodrug Ag | Method for prophylactic treatment of alzheimer's disease using inhibitors of glutaminyl cyclase and glutamate cyclases |
| EP1961416A1 (en) | 2003-05-05 | 2008-08-27 | Probiodrug AG | Medical use of inhibitors of glutaminyl and glutamate cyclases |
| WO2004098625A2 (en) | 2003-05-05 | 2004-11-18 | Probiodrug Ag | Medical use of inhibitors of glutaminyl and glutamate cyclases |
| US7560455B2 (en) | 2003-05-14 | 2009-07-14 | Merck & Co., Inc. | 3-Amino-4-phenylbutanoic acid derivatives as dipeptidyl peptidase inhibitors for the treatment or prevention of diabetes |
| US7332520B2 (en) | 2003-06-06 | 2008-02-19 | Merck & Co., Inc. | Fused indoles as dipeptidyl peptidase inhibitors for the treatment or prevention of diabetes |
| US7456204B2 (en) | 2003-06-17 | 2008-11-25 | Merck & Co., Inc. | Cyclohexylglycine derivatives as dipeptidyl peptidase inhibitors for the treatment or prevention of diabetes |
| US7718666B2 (en) | 2003-06-20 | 2010-05-18 | Hoffmann-La Roche Inc. | Pyrido [2,1-a] isoquinoline derivatives |
| US7122555B2 (en) | 2003-06-20 | 2006-10-17 | Hoffmann-La Roche Inc. | Pyrido [2,1-a] isoquinoline derivatives |
| US7259160B2 (en) | 2003-07-31 | 2007-08-21 | Merck & Co., Inc. | Hexahydrodiazepinones as dipeptidyl peptidase-IV inhibitors for the treatment or prevention of diabetes |
| EP1997489A1 (en) * | 2003-08-01 | 2008-12-03 | Bristol-Myers Squibb Company | Adamantylglycine derivatives as inhibitors of dipeptidyl peptidase IV for the treatment of diabetes |
| WO2005012249A3 (en) * | 2003-08-01 | 2005-05-06 | Bristol Myers Squibb Co | Adamantyglycine- based inhibitors of dipeptidyl peptidase iv for the treatment of diabetes |
| WO2005012249A2 (en) | 2003-08-01 | 2005-02-10 | Bristol-Myers Squibb Company | Adamantyglycine- based inhibitors of dipeptidyl peptidase iv for the treatment of diabetes |
| US7291599B2 (en) | 2003-08-12 | 2007-11-06 | Keyneurotek Ag | Use of inhibitors of enzymes having activities of amino peptidase N and/or dipeptidyl peptidase IV and of pharmaceutical preparations thereof for a therapy and prevention of chronical neurodegenerative diseases |
| EP1506776A1 (en) | 2003-08-12 | 2005-02-16 | KeyNeurotek AG | Use of enzyme inhibitors of the dipeptidylpeptidase IV and/or of the aminopeptidase n , and pharmaceutical preparations thereof for the prevention or therapy of neurodegenerative diseases |
| US7790736B2 (en) | 2003-08-13 | 2010-09-07 | Takeda Pharmaceutical Company Limited | Dipeptidyl peptidase inhibitors |
| US7678909B1 (en) | 2003-08-13 | 2010-03-16 | Takeda Pharmaceutical Company Limited | Dipeptidyl peptidase inhibitors |
| US7723344B2 (en) | 2003-08-13 | 2010-05-25 | Takeda San Diego, Inc. | Dipeptidyl peptidase inhibitors |
| US7169926B1 (en) | 2003-08-13 | 2007-01-30 | Takeda Pharmaceutical Company Limited | Dipeptidyl peptidase inhibitors |
| US7652021B2 (en) | 2003-08-29 | 2010-01-26 | Sanofi-Aventis | Compounds useful for DPP-IV enzyme inhibition |
| WO2005021536A2 (en) * | 2003-08-29 | 2005-03-10 | Sanofi-Aventis | Adamantane and azabicyclo-octane and nonane derivatives, process of their preparation and their use as dpp-iv inhibitors |
| AU2004268832B2 (en) * | 2003-08-29 | 2011-04-28 | Sanofi-Aventis | Adamantane and azabicyclo-octane and nonane derivatives, process of their preparation and their use as DPP-IV inhibitors |
| WO2005021536A3 (en) * | 2003-08-29 | 2005-10-13 | Sanofi Aventis | Adamantane and azabicyclo-octane and nonane derivatives, process of their preparation and their use as dpp-iv inhibitors |
| US7205409B2 (en) | 2003-09-04 | 2007-04-17 | Abbott Laboratories | Pharmaceutical compositions as inhibitors of dipeptidyl peptidase-IV (DPP-IV) |
| US7790734B2 (en) | 2003-09-08 | 2010-09-07 | Takeda Pharmaceutical Company Limited | Dipeptidyl peptidase inhibitors |
| EP2289498A1 (en) | 2003-10-15 | 2011-03-02 | Probiodrug AG | Use of inhibitors of glutaminyl clyclase |
| US7462599B2 (en) | 2003-10-15 | 2008-12-09 | Probiodrug Ag | Use of effectors of glutaminyl and glutamate cyclases |
| US7667044B2 (en) | 2003-11-03 | 2010-02-23 | Probiodrug Ag | Compounds for the treatment of neurological disorders |
| WO2005049027A2 (en) | 2003-11-03 | 2005-06-02 | Probiodrug Ag | Combinations useful for the treatment of neuronal disorders |
| EP2338490A2 (en) | 2003-11-03 | 2011-06-29 | Probiodrug AG | Combinations Useful for the Treatment of Neuronal Disorders |
| EP2839832A2 (en) | 2003-11-17 | 2015-02-25 | Novartis AG | Use of dipeptidyl peptidase IV inhibitors |
| JPWO2005053695A1 (en) * | 2003-12-04 | 2007-12-06 | エーザイ・アール・アンド・ディー・マネジメント株式会社 | Agents for preventing or treating multiple sclerosis |
| EP2165703A2 (en) | 2004-01-20 | 2010-03-24 | Novartis Pharma AG. | Direct compression formulation and process |
| EP3023095A1 (en) | 2004-01-20 | 2016-05-25 | Novartis AG | Direct compression formulation and process |
| EP3738585A1 (en) | 2004-01-20 | 2020-11-18 | Novartis Ag | Direct compression formulation and process |
| EP3366283A1 (en) | 2004-01-20 | 2018-08-29 | Novartis AG | Direct compression formulation and process |
| US8053465B2 (en) | 2004-02-05 | 2011-11-08 | Kyorin Pharmaceutical Co., Ltd. | Bicycloester derivative |
| US7304086B2 (en) | 2004-02-05 | 2007-12-04 | Probiodrug Ag | Inhibitors of glutaminyl cyclase |
| US7754757B2 (en) | 2004-02-05 | 2010-07-13 | Kyorin Pharmaceutical Co., Ltd. | Bicycloester derivative |
| WO2005079795A3 (en) * | 2004-02-20 | 2005-11-10 | Novartis Ag | Dpp-iv inhibitors for treating neurodegeneration and cognitive disorders |
| WO2005079795A2 (en) * | 2004-02-20 | 2005-09-01 | Novartis Ag | Dpp-iv inhibitors for treating neurodegeneration and cognitive disorders |
| US7732446B1 (en) | 2004-03-11 | 2010-06-08 | Takeda Pharmaceutical Company Limited | Dipeptidyl peptidase inhibitors |
| US8188275B2 (en) | 2004-03-15 | 2012-05-29 | Takeda Pharmaceutical Company Limited | Dipeptidyl peptidase inhibitors |
| US7906523B2 (en) | 2004-03-15 | 2011-03-15 | Takeda Pharmaceutical Company Limited | Dipeptidyl peptidase inhibitors |
| US8173663B2 (en) | 2004-03-15 | 2012-05-08 | Takeda Pharmaceutical Company Limited | Dipeptidyl peptidase inhibitors |
| US7781584B2 (en) | 2004-03-15 | 2010-08-24 | Takeda Pharmaceutical Company Limited | Dipeptidyl peptidase inhibitors |
| US8329900B2 (en) | 2004-03-15 | 2012-12-11 | Takeda Pharmaceutical Company Limited | Dipeptidyl peptidase inhibitors |
| US8288539B2 (en) | 2004-03-15 | 2012-10-16 | Takeda Pharmaceutical Company Limited | Dipeptidyl peptidase inhibitors |
| US7807689B2 (en) | 2004-03-15 | 2010-10-05 | Takeda Pharmaceutical Company Limited | Dipeptidyl peptidase inhibitors |
| US7687492B2 (en) | 2004-05-04 | 2010-03-30 | Merck Sharp & Dohme Corp. | 1,2,4-Oxadiazole derivatives as dipeptidyl peptidase-IV inhibitors for the treatment or prevention of diabetes |
| US7671073B2 (en) | 2004-05-18 | 2010-03-02 | Merck Sharp & Dohme Corp. | Cyclohexylalanine derivatives as dipeptidyl peptidase-IV inhibitors for the treatment or prevention of diabetes |
| US7687638B2 (en) | 2004-06-04 | 2010-03-30 | Takeda San Diego, Inc. | Dipeptidyl peptidase inhibitors |
| US7825242B2 (en) | 2004-07-16 | 2010-11-02 | Takeda Pharmaceutical Company Limted | Dipeptidyl peptidase inhibitors |
| US7872124B2 (en) | 2004-12-21 | 2011-01-18 | Takeda Pharmaceutical Company Limited | Dipeptidyl peptidase inhibitors |
| EP2116235A1 (en) | 2005-01-10 | 2009-11-11 | Arena Pharmaceuticals, Inc. | Combination therapy for the treatment of diabetes and conditions related thereto and for the treatment of conditions ameliorated by increasing a blood GLP-1 level |
| WO2007085895A3 (en) * | 2005-09-02 | 2008-04-17 | Ferring Bv | Fap inhibitors |
| EP1760076A1 (en) * | 2005-09-02 | 2007-03-07 | Ferring B.V. | FAP Inhibitors |
| WO2007085895A2 (en) * | 2005-09-02 | 2007-08-02 | Ferring B.V. | Fap inhibitors |
| US8183280B2 (en) | 2005-09-02 | 2012-05-22 | Vantia Limited | FAP inhibitors |
| US8906901B2 (en) | 2005-09-14 | 2014-12-09 | Takeda Pharmaceutical Company Limited | Administration of dipeptidyl peptidase inhibitors |
| US8222411B2 (en) | 2005-09-16 | 2012-07-17 | Takeda Pharmaceutical Company Limited | Dipeptidyl peptidase inhibitors |
| US8431540B2 (en) | 2005-12-23 | 2013-04-30 | Zealand Pharma A/S | Modified lysine-mimetic compounds |
| WO2007072083A1 (en) | 2005-12-23 | 2007-06-28 | Prosidion Limited | Treatment of type 2 diabetes with a combination of dpiv inhibitor and metformin or thiazolidinedione |
| US7622496B2 (en) | 2005-12-23 | 2009-11-24 | Zealand Pharma A/S | Modified lysine-mimetic compounds |
| KR100792275B1 (en) * | 2006-02-22 | 2008-01-08 | 영진약품공업주식회사 | Cyclic hydrazide derivatives having ?-amino group, its pharmaceutical acceptable salts and preparation process thereof |
| US7915427B2 (en) | 2006-03-08 | 2011-03-29 | Kyorin Pharmaceuticals Co., Ltd. | Process for producing aminoacetyl pyrrolidine carbonitrile derivative and intermediate for production thereof |
| US7960384B2 (en) | 2006-03-28 | 2011-06-14 | Takeda Pharmaceutical Company Limited | Dipeptidyl peptidase inhibitors |
| EP2253311A2 (en) | 2006-04-11 | 2010-11-24 | Arena Pharmaceuticals, Inc. | Use of GPR119 receptor agonists for increasing bone mass and for treating osteoporosis, as well as combination therapy relating thereto |
| WO2007120702A2 (en) | 2006-04-11 | 2007-10-25 | Arena Pharmaceuticals, Inc. | Use of gpr119 receptor agonists for increasing bone mass and for treating osteoporosis, and combination therapy relating thereto |
| US7728146B2 (en) | 2006-04-12 | 2010-06-01 | Probiodrug Ag | Enzyme inhibitors |
| US8324383B2 (en) | 2006-09-13 | 2012-12-04 | Takeda Pharmaceutical Company Limited | Methods of making polymorphs of benzoate salt of 2-[[6-[(3R)-3-amino-1-piperidinyl]-3,4-dihydro-3-methyl-2,4-dioxo-1(2H)-pyrimidinyl]methyl]-benzonitrile |
| WO2008055945A1 (en) | 2006-11-09 | 2008-05-15 | Probiodrug Ag | 3-hydr0xy-1,5-dihydr0-pyrr0l-2-one derivatives as inhibitors of glutaminyl cyclase for the treatment of ulcer, cancer and other diseases |
| US8084605B2 (en) | 2006-11-29 | 2011-12-27 | Kelly Ron C | Polymorphs of succinate salt of 2-[6-(3-amino-piperidin-1-yl)-3-methyl-2,4-dioxo-3,4-dihydro-2H-pyrimidin-1-ylmethy]-4-fluor-benzonitrile and methods of use therefor |
| WO2008065141A1 (en) | 2006-11-30 | 2008-06-05 | Probiodrug Ag | Novel inhibitors of glutaminyl cyclase |
| US9469609B2 (en) | 2006-12-21 | 2016-10-18 | Zealand Pharma A/S | Synthesis of pyrrolidine compounds |
| US8927590B2 (en) | 2006-12-21 | 2015-01-06 | Zealand Pharma A/S | Synthesis of pyrrolidine compounds |
| US8093236B2 (en) | 2007-03-13 | 2012-01-10 | Takeda Pharmaceuticals Company Limited | Weekly administration of dipeptidyl peptidase inhibitors |
| US8143427B2 (en) | 2007-03-22 | 2012-03-27 | Kyorin Pharmaceutical Co., Ltd. | Method for producing aminoacetylpyrrolidinecarbonitrile derivative |
| EP2865670A1 (en) | 2007-04-18 | 2015-04-29 | Probiodrug AG | Thiourea derivatives as glutaminyl cyclase inhibitors |
| US8883714B2 (en) | 2008-04-07 | 2014-11-11 | Arena Pharmaceuticals, Inc. | Pharmaceutical compositions comprising GPR119 agonists which act as peptide YY (PYY) secretagogues |
| WO2011005929A1 (en) | 2009-07-09 | 2011-01-13 | Arena Pharmaceuticals, Inc. | Piperidine derivative and its use for the treatment of diabets and obesity |
| WO2011029920A1 (en) | 2009-09-11 | 2011-03-17 | Probiodrug Ag | Heterocylcic derivatives as inhibitors of glutaminyl cyclase |
| WO2011107530A2 (en) | 2010-03-03 | 2011-09-09 | Probiodrug Ag | Novel inhibitors |
| WO2011110613A1 (en) | 2010-03-10 | 2011-09-15 | Probiodrug Ag | Heterocyclic inhibitors of glutaminyl cyclase (qc, ec 2.3.2.5) |
| WO2011127051A1 (en) | 2010-04-06 | 2011-10-13 | Arena Pharmaceuticals, Inc. | Modulators of the gpr119 receptor and the treatment of disorders related thereto |
| WO2011131748A2 (en) | 2010-04-21 | 2011-10-27 | Probiodrug Ag | Novel inhibitors |
| WO2012040279A1 (en) | 2010-09-22 | 2012-03-29 | Arena Pharmaceuticals, Inc. | Modulators of the gpr119 receptor and the treatment of disorders related thereto |
| EP3323818A1 (en) | 2010-09-22 | 2018-05-23 | Arena Pharmaceuticals, Inc. | Modulators of the gpr119 receptor and the treatment of disorders related thereto |
| WO2012123563A1 (en) | 2011-03-16 | 2012-09-20 | Probiodrug Ag | Benz imidazole derivatives as inhibitors of glutaminyl cyclase |
| WO2012135570A1 (en) | 2011-04-01 | 2012-10-04 | Arena Pharmaceuticals, Inc. | Modulators of the gpr119 receptor and the treatment of disorders related thereto |
| WO2012145361A1 (en) | 2011-04-19 | 2012-10-26 | Arena Pharmaceuticals, Inc. | Modulators of the gpr119 receptor and the treatment of disorders related thereto |
| WO2012145603A1 (en) | 2011-04-22 | 2012-10-26 | Arena Pharmaceuticals, Inc. | Modulators of the gpr119 receptor and the treatment of disorders related thereto |
| WO2012145604A1 (en) | 2011-04-22 | 2012-10-26 | Arena Pharmaceuticals, Inc. | Modulators of the gpr119 receptor and the treatment of disorders related thereto |
| WO2012170702A1 (en) | 2011-06-08 | 2012-12-13 | Arena Pharmaceuticals, Inc. | Modulators of the gpr119 receptor and the treatment of disorders related thereto |
| WO2013055910A1 (en) | 2011-10-12 | 2013-04-18 | Arena Pharmaceuticals, Inc. | Modulators of the gpr119 receptor and the treatment of disorders related thereto |
| US9115082B2 (en) | 2012-01-18 | 2015-08-25 | Catherine Yang | Dipeptidyl-peptidase-IV inhibitors for treatment of type 2 diabetes complex with hypertension |
| WO2014074668A1 (en) | 2012-11-08 | 2014-05-15 | Arena Pharmaceuticals, Inc. | Modulators of gpr119 and the treatment of disorders related thereto |
| WO2014113750A1 (en) | 2013-01-18 | 2014-07-24 | Dnj Pharma, Inc. | Novel ddp-iv inhibitors |
| EP4000630A1 (en) | 2014-09-03 | 2022-05-25 | Invex Therapeutics Ltd | Elevated intracranial pressure treatment |
| US10555929B2 (en) | 2015-03-09 | 2020-02-11 | Coherus Biosciences, Inc. | Methods for the treatment of nonalcoholic fatty liver disease and/or lipodystrophy |
| US10772865B2 (en) | 2015-03-09 | 2020-09-15 | Coherus Biosciences, Inc. | Methods for the treatment of nonalcoholic fatty liver disease and/or lipodystrophy |
| US11400072B2 (en) | 2015-03-09 | 2022-08-02 | Coherus Biosciences, Inc. | Methods for the treatment of nonalcoholic fatty liver disease and/or lipodystrophy |
| WO2018162722A1 (en) | 2017-03-09 | 2018-09-13 | Deutsches Institut Für Ernährungsforschung Potsdam-Rehbrücke | Dpp-4 inhibitors for use in treating bone fractures |
| US11253508B2 (en) | 2017-04-03 | 2022-02-22 | Coherus Biosciences, Inc. | PPARy agonist for treatment of progressive supranuclear palsy |
| US11324799B2 (en) | 2017-05-05 | 2022-05-10 | Zealand Pharma A/S | Gap junction intercellular communication modulators and their use for the treatment of diabetic eye disease |
| EP3461819A1 (en) | 2017-09-29 | 2019-04-03 | Probiodrug AG | Inhibitors of glutaminyl cyclase |
Also Published As
| Publication number | Publication date |
|---|---|
| CA2390231A1 (en) | 2001-05-17 |
| AU1916401A (en) | 2001-06-06 |
| JP2003535034A (en) | 2003-11-25 |
| EP1228061A1 (en) | 2002-08-07 |
| EP1228061A4 (en) | 2004-12-15 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP1228061A1 (en) | Dipeptidyl peptidase iv inhibitors and methods of making and using dipeptidyl peptidase iv inhibitors | |
| US20040152745A1 (en) | Dipeptidyl peptidase IV inhibitors and methods of making and using dipeptidyl peptidase IV inhibitors | |
| JP6892218B2 (en) | How to treat diseases caused by drug delivery conjugates and PSMA-expressing cells | |
| US7553830B2 (en) | Compositions for the delivery of negatively charged molecules | |
| TW201034690A (en) | Technetium-and rhenium-bis (heteroaryl) complexes and methods of use thereof for inhibiting PSMA | |
| JP2009500423A (en) | Fibroblast activation protein alpha inhibitor | |
| KR20000035925A (en) | Phosphinic acid amides as matrix metalloprotease inhibitors | |
| CZ2004973A3 (en) | Glutaminyl based DPIV inhibitors | |
| IL139862A (en) | Thiazolidine or pyrrolidine | |
| JP2000508319A (en) | αvβ3 antagonist | |
| CN102159553A (en) | Targeted nitroxide agents | |
| JP2018048178A (en) | Oxabicycloheptanes, and oxabicycloheptanes for treatment of reperfusion injury | |
| AU2014203872A1 (en) | Use of fatty acid niacin conjugates for treating diseases | |
| JP3832229B2 (en) | Phenylethenesulfonamide derivative-containing medicine | |
| JP2021521106A (en) | HSP90 targeted conjugate and its formulation | |
| US20130338372A1 (en) | Substituted Imidazoline Compounds | |
| JP2004026678A (en) | Therapeutic agent for type 2 diabetes | |
| JP2016537353A (en) | Fatty acid niacin complex | |
| AU3171293A (en) | Use of renin inhibitors for the treatment of glaucoma | |
| US11136295B2 (en) | D-amphetamine compounds, compositions, and processes for making and using the same | |
| KR20010083122A (en) | Novel remedies for allergic diseases | |
| JP2003503476A (en) | Cyclic amide derivative | |
| EP3607948A1 (en) | Tissue transglutaminase modulators for medicinal use | |
| CA3123368A1 (en) | Mitochondria-targeting peptides | |
| MXPA02004679A (en) | Dipeptidyl peptidase iv inhibitors and methods of making and using dipeptidyl peptidase iv inhibitors |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): AE AG AL AM AT AU AZ BA BB BG BR BY BZ CA CH CN CR CU CZ DE DK DM DZ EE ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MA MD MG MK MN MW MX MZ NO NZ PL PT RO RU SD SE SG SI SK SL TJ TM TR TT TZ UA UG US UZ VN YU ZA ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): GH GM KE LS MW MZ SD SL SZ TZ UG ZW AM AZ BY KG KZ MD RU TJ TM AT BE CH CY DE DK ES FI FR GB GR IE IT LU MC NL PT SE TR BF BJ CF CG CI CM GA GN GW ML MR NE SN TD TG |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 2390231 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 19164/01 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2000982093 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: PA/a/2002/004679 Country of ref document: MX |
|
| ENP | Entry into the national phase |
Ref country code: JP Ref document number: 2001 536541 Kind code of ref document: A Format of ref document f/p: F |
|
| WWP | Wipo information: published in national office |
Ref document number: 2000982093 Country of ref document: EP |
|
| REG | Reference to national code |
Ref country code: DE Ref legal event code: 8642 |
|
| WWW | Wipo information: withdrawn in national office |
Ref document number: 2000982093 Country of ref document: EP |
|
| DPE2 | Request for preliminary examination filed before expiration of 19th month from priority date (pct application filed from 20040101) |