WO2001012187A2 - Benzoic acid derivatives and their use as ppar receptor agonists - Google Patents
Benzoic acid derivatives and their use as ppar receptor agonists Download PDFInfo
- Publication number
- WO2001012187A2 WO2001012187A2 PCT/GB2000/003140 GB0003140W WO0112187A2 WO 2001012187 A2 WO2001012187 A2 WO 2001012187A2 GB 0003140 W GB0003140 W GB 0003140W WO 0112187 A2 WO0112187 A2 WO 0112187A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- optionally substituted
- formula
- compound
- group
- alkyl
- Prior art date
Links
- QUIZHBVFOHDTRS-UHFFFAOYSA-N CCOC(c1c(C[n](ccc2c3)c2ccc3OCC(NCc2ccccc2)=O)cccc1)=O Chemical compound CCOC(c1c(C[n](ccc2c3)c2ccc3OCC(NCc2ccccc2)=O)cccc1)=O QUIZHBVFOHDTRS-UHFFFAOYSA-N 0.000 description 1
- IJZMXROFNOQHLW-UHFFFAOYSA-N CCOC(c1ccccc1C[n](ccc1c2)c1ccc2-c(cc1)ccc1C(O)=O)=O Chemical compound CCOC(c1ccccc1C[n](ccc1c2)c1ccc2-c(cc1)ccc1C(O)=O)=O IJZMXROFNOQHLW-UHFFFAOYSA-N 0.000 description 1
- JXFOKIHHFHNEMY-UHFFFAOYSA-N COC(c1c(C[n]2c3ccc(CNC(NCc4ccccc4)=O)cc3cc2)cccc1)=O Chemical compound COC(c1c(C[n]2c3ccc(CNC(NCc4ccccc4)=O)cc3cc2)cccc1)=O JXFOKIHHFHNEMY-UHFFFAOYSA-N 0.000 description 1
- TXHASZTWOTYMAR-UHFFFAOYSA-N COC(c1ccccc1C[n]1c2ccc(CN)cc2cc1)=O Chemical compound COC(c1ccccc1C[n]1c2ccc(CN)cc2cc1)=O TXHASZTWOTYMAR-UHFFFAOYSA-N 0.000 description 1
- XRLWBRGIVHCOFT-UHFFFAOYSA-N COC(c1ccccc1C[n]1c2ccc(CNC(c(cc3)ccc3-c3ccccc3)=O)cc2cc1)=O Chemical compound COC(c1ccccc1C[n]1c2ccc(CNC(c(cc3)ccc3-c3ccccc3)=O)cc2cc1)=O XRLWBRGIVHCOFT-UHFFFAOYSA-N 0.000 description 1
- USRFISAWFLLDKQ-UHFFFAOYSA-N Cc(c1c2)c[n](Cc(cccc3)c3C(O)=O)c1ccc2OCc1nc(cccc2)c2cc1 Chemical compound Cc(c1c2)c[n](Cc(cccc3)c3C(O)=O)c1ccc2OCc1nc(cccc2)c2cc1 USRFISAWFLLDKQ-UHFFFAOYSA-N 0.000 description 1
- 0 Cc1c[n](Cc2c(*)cccc2)c2cc(*)c(C3C*CC3)c(*)c12 Chemical compound Cc1c[n](Cc2c(*)cccc2)c2cc(*)c(C3C*CC3)c(*)c12 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D215/00—Heterocyclic compounds containing quinoline or hydrogenated quinoline ring systems
- C07D215/02—Heterocyclic compounds containing quinoline or hydrogenated quinoline ring systems having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen atoms or carbon atoms directly attached to the ring nitrogen atom
- C07D215/16—Heterocyclic compounds containing quinoline or hydrogenated quinoline ring systems having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen atoms or carbon atoms directly attached to the ring nitrogen atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D215/20—Oxygen atoms
- C07D215/22—Oxygen atoms attached in position 2 or 4
- C07D215/227—Oxygen atoms attached in position 2 or 4 only one oxygen atom which is attached in position 2
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/47—Quinolines; Isoquinolines
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/40—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with one nitrogen as the only ring hetero atom, e.g. sulpiride, succinimide, tolmetin, buflomedil
- A61K31/403—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with one nitrogen as the only ring hetero atom, e.g. sulpiride, succinimide, tolmetin, buflomedil condensed with carbocyclic rings, e.g. carbazole
- A61K31/404—Indoles, e.g. pindolol
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/4427—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems
- A61K31/4439—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems containing a five-membered ring with nitrogen as a ring hetero atom, e.g. omeprazole
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/47—Quinolines; Isoquinolines
- A61K31/4704—2-Quinolinones, e.g. carbostyril
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/47—Quinolines; Isoquinolines
- A61K31/4709—Non-condensed quinolines and containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D209/00—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D209/02—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom condensed with one carbocyclic ring
- C07D209/04—Indoles; Hydrogenated indoles
- C07D209/08—Indoles; Hydrogenated indoles with only hydrogen atoms or radicals containing only hydrogen and carbon atoms, directly attached to carbon atoms of the hetero ring
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D215/00—Heterocyclic compounds containing quinoline or hydrogenated quinoline ring systems
- C07D215/02—Heterocyclic compounds containing quinoline or hydrogenated quinoline ring systems having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen atoms or carbon atoms directly attached to the ring nitrogen atom
- C07D215/16—Heterocyclic compounds containing quinoline or hydrogenated quinoline ring systems having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen atoms or carbon atoms directly attached to the ring nitrogen atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D215/20—Oxygen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D231/00—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings
- C07D231/02—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings not condensed with other rings
- C07D231/10—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members
- C07D231/12—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members with only hydrogen atoms, hydrocarbon or substituted hydrocarbon radicals, directly attached to ring carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D233/00—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings
- C07D233/54—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings having two double bonds between ring members or between ring members and non-ring members
- C07D233/56—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings having two double bonds between ring members or between ring members and non-ring members with only hydrogen atoms or radicals containing only hydrogen and carbon atoms, attached to ring carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D249/00—Heterocyclic compounds containing five-membered rings having three nitrogen atoms as the only ring hetero atoms
- C07D249/02—Heterocyclic compounds containing five-membered rings having three nitrogen atoms as the only ring hetero atoms not condensed with other rings
- C07D249/08—1,2,4-Triazoles; Hydrogenated 1,2,4-triazoles
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/06—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a carbon chain containing only aliphatic carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/02—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings
- C07D405/04—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/02—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings
- C07D405/12—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D409/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms
- C07D409/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings
- C07D409/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings
- C07D413/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings
- C07D417/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings linked by a chain containing hetero atoms as chain links
Abstract
Description
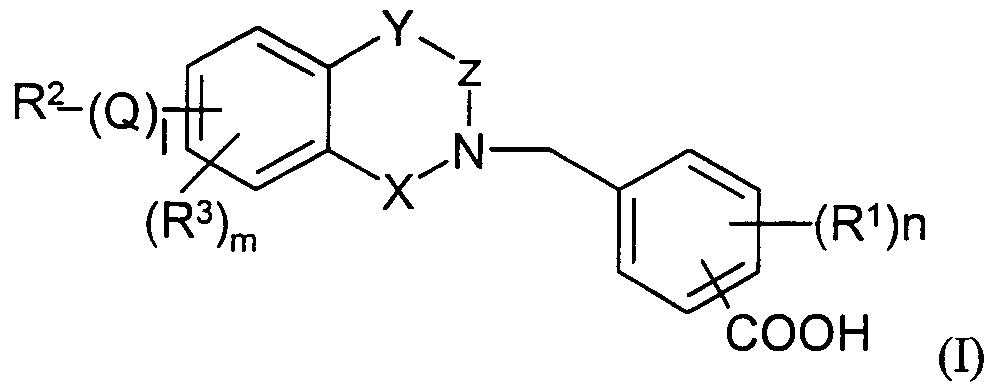





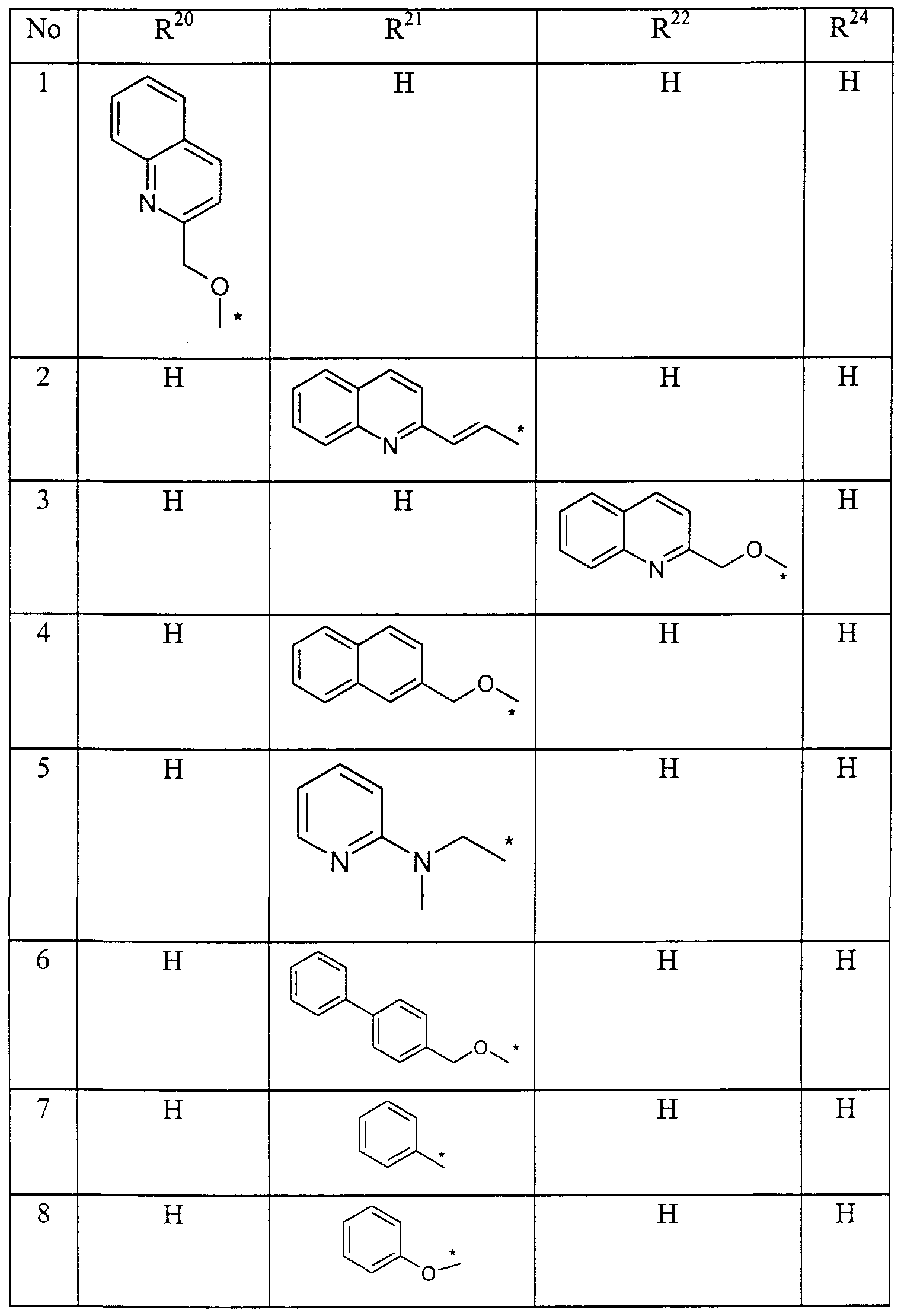







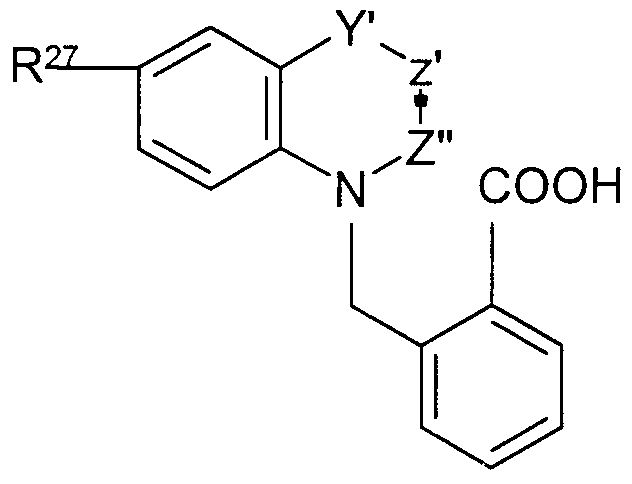















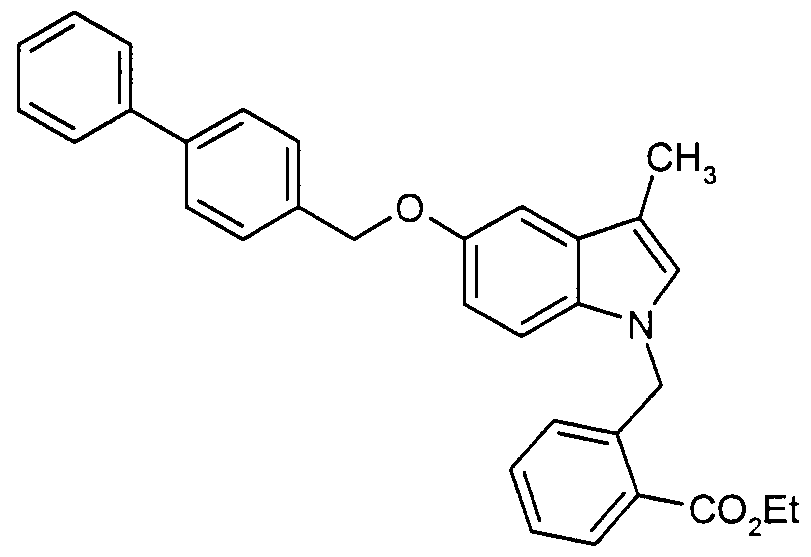

































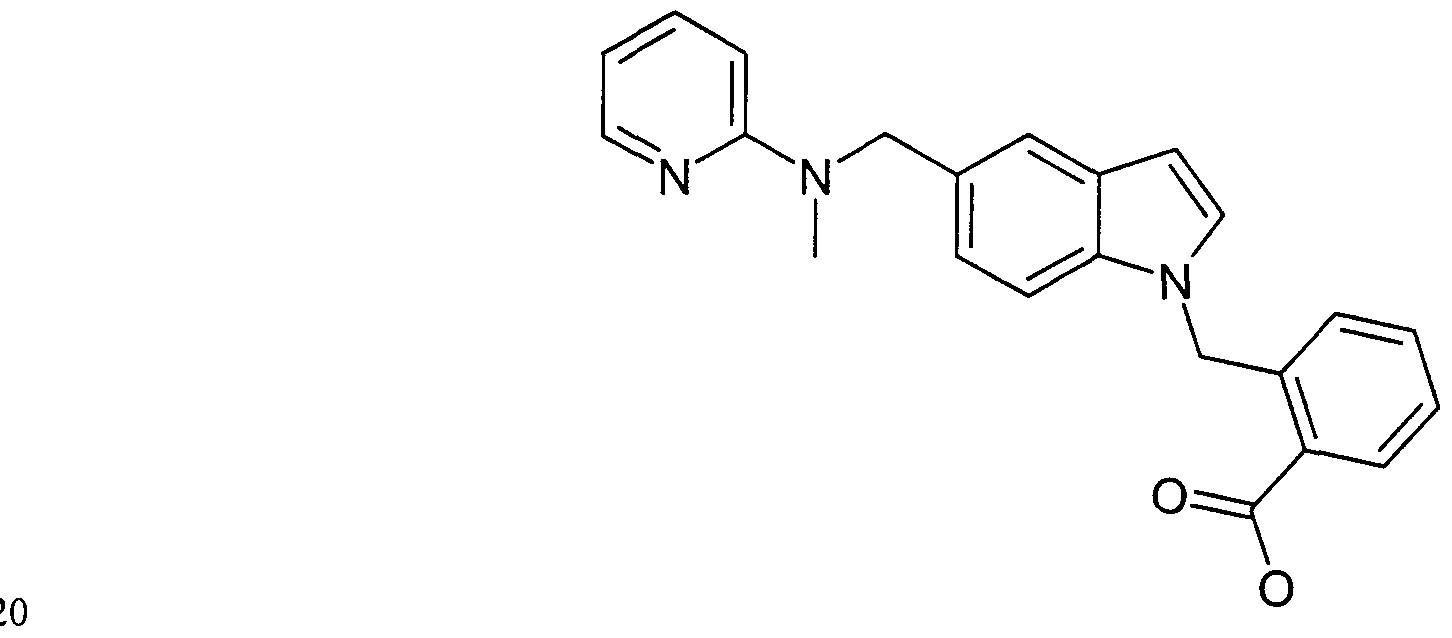














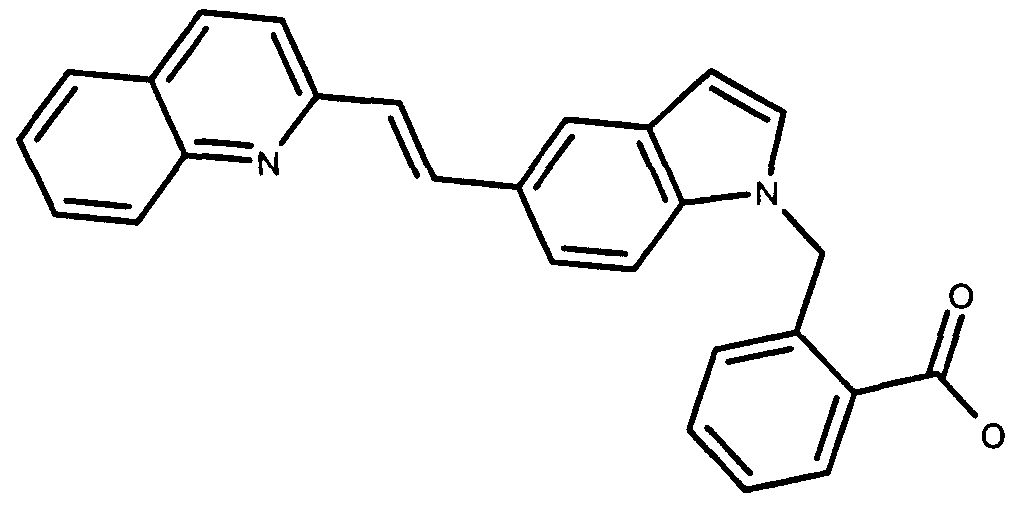


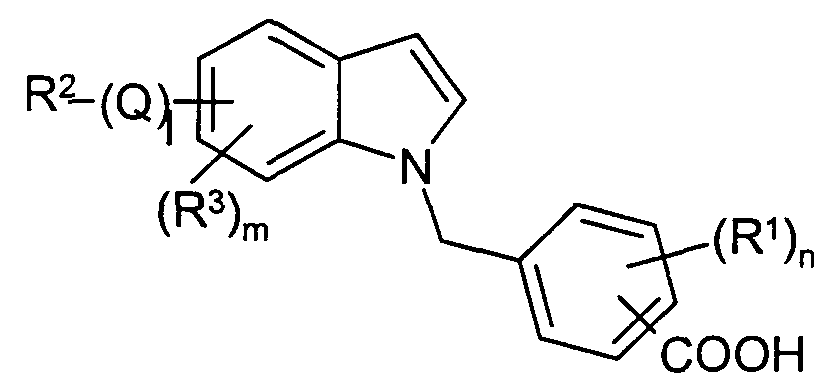
Claims



Priority Applications (10)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CA002380775A CA2380775A1 (en) | 1999-08-18 | 2000-08-14 | Benzoic acid derivatives and their use as ppar receptor agonists |
| IL14782100A IL147821A0 (en) | 1999-08-18 | 2000-08-14 | Benzoic acid derivatives and their use as ppar receptor agonists |
| BR0013368-0A BR0013368A (en) | 1999-08-18 | 2000-08-14 | Use of a compound, compound, and pharmaceutical composition |
| MXPA02001598A MXPA02001598A (en) | 1999-08-18 | 2000-08-14 | Chemical compounds. |
| JP2001516533A JP2003507327A (en) | 1999-08-18 | 2000-08-14 | Chemical compound |
| EP00953320A EP1210343A2 (en) | 1999-08-18 | 2000-08-14 | Benzoic acid derivatives and their use as ppar receptor agonists |
| KR1020027002019A KR20020020817A (en) | 1999-08-18 | 2000-08-14 | Chemical Compounds |
| NZ517059A NZ517059A (en) | 1999-08-18 | 2000-08-14 | Indole derivatives of benzoic acid which act as peroxisome proliferator activated receptor (PPAR) |
| AU65834/00A AU6583400A (en) | 1999-08-18 | 2000-08-14 | Chemical compounds |
| NO20020765A NO20020765L (en) | 1999-08-18 | 2002-02-15 | Chemical connections |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| GB9919411.0 | 1999-08-18 | ||
| GBGB9919411.0A GB9919411D0 (en) | 1999-08-18 | 1999-08-18 | Chemical compounds |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2001012187A2 true WO2001012187A2 (en) | 2001-02-22 |
| WO2001012187A3 WO2001012187A3 (en) | 2001-06-07 |
Family
ID=10859283
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/GB2000/003140 WO2001012187A2 (en) | 1999-08-18 | 2000-08-14 | Benzoic acid derivatives and their use as ppar receptor agonists |
Country Status (14)
| Country | Link |
|---|---|
| EP (1) | EP1210343A2 (en) |
| JP (1) | JP2003507327A (en) |
| KR (1) | KR20020020817A (en) |
| CN (1) | CN1379774A (en) |
| AU (1) | AU6583400A (en) |
| BR (1) | BR0013368A (en) |
| CA (1) | CA2380775A1 (en) |
| GB (1) | GB9919411D0 (en) |
| IL (1) | IL147821A0 (en) |
| MX (1) | MXPA02001598A (en) |
| NO (1) | NO20020765L (en) |
| NZ (1) | NZ517059A (en) |
| WO (1) | WO2001012187A2 (en) |
| ZA (1) | ZA200200669B (en) |
Cited By (53)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2002044149A1 (en) * | 2000-11-28 | 2002-06-06 | F. Hoffmann-La Roche Ag | Indole and dihydroindole derivatives |
| WO2003035602A1 (en) * | 2001-10-25 | 2003-05-01 | Sankyo Company, Limited | Lipid modulators |
| WO2003066581A1 (en) * | 2002-02-05 | 2003-08-14 | Eli Lilly And Company | Urea linker derivatives for use as ppar modulators |
| WO2004000295A1 (en) * | 2002-06-20 | 2003-12-31 | Astrazeneca Ab | Benzoic acid derivatives as modulators of ppar alpha and gamma |
| WO2004096767A1 (en) * | 2003-04-25 | 2004-11-11 | H. Lundbeck A/S | Sustituted indoline and indole derivatives |
| US6943169B2 (en) | 2003-03-11 | 2005-09-13 | Roche Palo Alto Llc | Quinolinone derivatives and uses thereof |
| US7056943B2 (en) | 2002-12-10 | 2006-06-06 | Wyeth | Substituted indole oxo-acetyl amino acetic acid derivatives as inhibitors of plasminogen activator inhibitor-1 (PAI-1) |
| US7074817B2 (en) | 2001-06-20 | 2006-07-11 | Wyeth | Substituted indole acid derivatives as inhibitors of plasminogen activator inhibitor-1 (PAI-1) |
| US7078429B2 (en) | 2002-12-10 | 2006-07-18 | Wyeth | Substituted 3-carbonyl-1H-indol-1-yl acetic acid derivatives as inhibitors of plasminogen activator inhibitor-1 (PAI-1) |
| US7101903B2 (en) | 2002-12-10 | 2006-09-05 | Wyeth | Substituted dihydropyrano indole-3,4-dione derivatives as inhibitiors of plasminogen activator inhibitor-1 (PAI-1) |
| US7129264B2 (en) | 2003-04-16 | 2006-10-31 | Bristol-Myers Squibb Company | Biarylmethyl indolines and indoles as antithromboembolic agents |
| US7163954B2 (en) | 2003-09-25 | 2007-01-16 | Wyeth | Substituted naphthyl benzothiophene acids |
| WO2007063418A2 (en) * | 2005-04-13 | 2007-06-07 | Neuraxon, Inc. | Substituted indole compounds having nos inhibitory activity |
| US7259182B2 (en) | 2002-12-10 | 2007-08-21 | Wyeth | Aryl, aryloxy, and aklyloxy substituted 1H-indol-3-yl glyoxylic acid derivatives as inhibitors of plasminogen activator inhibitor-1 (PAI-1) |
| US7268159B2 (en) | 2003-09-25 | 2007-09-11 | Wyeth | Substituted indoles |
| US7291639B2 (en) | 2001-06-20 | 2007-11-06 | Wyeth | Aryloxy-acetic acid compounds useful as inhibitors of plasminogen activator inhibitor-1 (PAI-1) |
| WO2008020302A2 (en) * | 2006-08-17 | 2008-02-21 | Pfizer Products Inc. | Heteroaromatic quinoline-based compounds as phosphodiesterase (pde) inhibitors |
| US7348351B2 (en) | 2002-12-10 | 2008-03-25 | Wyeth | Substituted 3-alkyl and 3-arylalkyl 1H-indol-1yl acetic acid derivatives as inhibitors of plasminogen activator inhibitor-1 (PAI-1) |
| US7351726B2 (en) | 2003-09-25 | 2008-04-01 | Wyeth | Substituted oxadiazolidinediones |
| US7351730B2 (en) | 2001-06-20 | 2008-04-01 | Wyeth | Substituted naphthyl indole derivatives as inhibitors of plasminogen activator inhibitor type-1 (PAI-1) |
| US7355069B2 (en) | 2002-06-20 | 2008-04-08 | Astrazeneca Ab | Ortho-substituted benzoic acid derivatives for the treatment of insulin resistance |
| EP1911462A2 (en) | 2001-01-26 | 2008-04-16 | Schering Corporation | Compositions comprising a sterol absorption inhibitor |
| WO2008109336A1 (en) | 2007-03-01 | 2008-09-12 | Janssen Pharmaceutica N.V. | Tetrahydroisoquinoline compounds as modulators of the histamine h3 receptor |
| US7442805B2 (en) | 2003-09-25 | 2008-10-28 | Wyeth | Substituted sulfonamide-indoles |
| US7446201B2 (en) | 2003-09-25 | 2008-11-04 | Wyeth | Substituted heteroaryl benzofuran acids |
| WO2009005998A1 (en) * | 2007-07-02 | 2009-01-08 | Smithkline Beecham Corporation | Farnesoid x receptor agonists |
| FR2921366A1 (en) * | 2007-09-26 | 2009-03-27 | Servier Lab | New heterocyclic compounds are aldose reductase inhibitors useful to treat e.g. hyperglycemia, dyslipidemia, non-insulin-dependant type II diabetes, syndrome X, coronary artery diseases, glomerulonephritis and psoriasis |
| US7582773B2 (en) | 2003-09-25 | 2009-09-01 | Wyeth | Substituted phenyl indoles |
| WO2009100294A3 (en) * | 2008-02-07 | 2009-11-12 | Abbott Laboratories | Amide derivatives as positive allosteric modulators and methods of use thereof |
| US7683091B2 (en) | 2005-08-17 | 2010-03-23 | Wyeth | Substituted indoles and methods of their use |
| US7749999B2 (en) | 2003-09-11 | 2010-07-06 | Itherx Pharmaceuticals, Inc. | Alpha-ketoamides and derivatives thereof |
| US7749995B2 (en) | 2006-05-11 | 2010-07-06 | Janssen Pharmaceutica Nv | 3,4-dihydro-2h-benzo[1,4]oxazine and thiazine derivatives as CETP inhibitors |
| US7754747B2 (en) | 2004-08-23 | 2010-07-13 | Wyeth Llc | Oxazolo-naphthyl acids |
| US7803835B2 (en) | 2003-09-25 | 2010-09-28 | Wyeth Llc | Substituted acetic acid derivatives |
| US7928238B2 (en) | 2006-05-11 | 2011-04-19 | Janssen Pharmaceutica Nv | 1,2,3,4-tetrahydro-quinoline derivatives as CETP inhibitors |
| US7989447B2 (en) | 2006-04-13 | 2011-08-02 | Neuraxon, Inc. | 1,5 and 3,6-substituted indole compounds having NOS inhibitory activity |
| US8093265B2 (en) | 2007-03-09 | 2012-01-10 | Renovis, Inc. | Bicycloheteroaryl compounds as P2X7 modulators and uses thereof |
| WO2012027331A1 (en) | 2010-08-27 | 2012-03-01 | Ironwood Pharmaceuticals, Inc. | Compositions and methods for treating or preventing metabolic syndrome and related diseases and disorders |
| WO2012064267A1 (en) | 2010-11-08 | 2012-05-18 | Albireo Ab | A pharmaceutical combination comprising an ibat inhibitor and a bile acid binder |
| WO2012064266A1 (en) | 2010-11-08 | 2012-05-18 | Albireo Ab | Ibat inhibitors for the treatment of liver diseases |
| US20120309769A1 (en) * | 2011-06-06 | 2012-12-06 | Scripps Research Institute, The | N-benzylindole modulators of pparg |
| US20130184463A1 (en) * | 2011-12-21 | 2013-07-18 | Allergan, Inc. | Compounds acting at multiple prostaglandin receptors giving a general anti-inflammatory response |
| US8673909B2 (en) | 2007-11-16 | 2014-03-18 | Neuraxon, Inc. | Indole compounds and methods for treating visceral pain |
| US8686002B2 (en) | 2005-08-21 | 2014-04-01 | AbbVie Deutschland GmbH & Co. KG | Heterocyclic compounds and their use as binding partners for 5-HT5 receptors |
| US8957093B2 (en) | 2011-06-06 | 2015-02-17 | The Scripps Research Institute | N-biphenylmethylindole modulators of PPARG |
| EP2736330A4 (en) * | 2011-07-29 | 2015-05-27 | Tempero Pharmaceuticals Inc | Compounds and methods |
| EP2844247A4 (en) * | 2012-04-20 | 2015-11-25 | Anderson Gaweco | Ror modulators and their uses |
| US9309227B2 (en) | 2011-11-22 | 2016-04-12 | The Scripps Research Institute | N-biphenylmethylbenzimidazole modulators of PPARG |
| US9708268B2 (en) | 2012-04-30 | 2017-07-18 | Innov17 Llc | ROR modulators and their uses |
| US10016394B2 (en) | 2014-04-16 | 2018-07-10 | The Scripps Research Institute | PPARG modulators for treatment of osteoporosis |
| WO2018214959A1 (en) * | 2017-05-26 | 2018-11-29 | 南京明德新药研发股份有限公司 | Lactam compound as fxr receptor agonist |
| WO2020045982A1 (en) * | 2018-08-29 | 2020-03-05 | 숙명여자대학교산학협력단 | SUBSTITUTED INDOLE DERIVATIVE, PREPARATION METHOD FOR SAME, AND PHARMACEUTICAL COMPOSITION COMPRISING SAME AS EFFECTIVE COMPONENT FOR PREVENTING OR TREATING DISEASES ASSOCIATED WITH PPARα, PPARγ, AND PPARδ |
| RU2776052C2 (en) * | 2017-05-26 | 2022-07-12 | СиЭсПиСи ЧЖУНЦИ ФАРМАСЬЮТИКАЛ ТЕКНОЛОДЖИ (ШИЦЗЯЧЖУАН) КО., ЛТД. | Lactam compound as fxr receptor agonist |
Families Citing this family (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6495949B1 (en) | 1999-11-03 | 2002-12-17 | Orion Electric Co., Ltd. | Electron tube cathode |
| DE10308352A1 (en) * | 2003-02-27 | 2004-09-09 | Aventis Pharma Deutschland Gmbh | Branched side chain arylcycloalkyl derivatives, process for their preparation and their use as medicaments |
| ATE399156T1 (en) * | 2004-10-27 | 2008-07-15 | Hoffmann La Roche | NEW INDOLE OR BENZIMIDAZOLE DERIVATIVES |
| US7432255B2 (en) * | 2006-05-16 | 2008-10-07 | Hoffmann-La Roche Inc. | 1H-indol-5-yl-piperazin-1-yl-methanone derivatives |
| CN104788358A (en) * | 2014-01-20 | 2015-07-22 | 中国科学院上海药物研究所 | N-(3-fluoro-4-chlorobenzyl)indole derivative and use thereof |
| KR101585605B1 (en) * | 2014-03-20 | 2016-01-21 | 현대약품 주식회사 | Compounds that binding with PPARG(Peroxisome Proliferator Activated Receptor-Gamma) but not act as an agonist and pharmaceutical composition for diseases related with PPARG containing the same as an active ingredient |
| CN107176914B (en) * | 2016-03-09 | 2022-06-28 | 浙江旭晨医药科技有限公司 | GVS series compound and its use |
| WO2020048547A1 (en) * | 2018-09-07 | 2020-03-12 | 南京明德新药研发有限公司 | Tricyclic furan-substituted piperidinedione compound |
Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0179619A1 (en) * | 1984-10-19 | 1986-04-30 | Ici Americas Inc. | Heterocyclic amides |
| US4894386A (en) * | 1987-04-15 | 1990-01-16 | Ici Americas Inc. | Aliphatic carboxamides |
| WO1997005114A1 (en) * | 1995-07-31 | 1997-02-13 | Laboratorios Menarini S.A. | Quinolone sulfonimides having leukotriene-antagonistic action |
| EP0780389A1 (en) * | 1995-12-22 | 1997-06-25 | Tobishi Pharmaceutical Co., Ltd. | Thiazolidinedione derivatives, process for their preparation and pharmaceutical compositions containing them |
| WO1998051667A1 (en) * | 1997-05-16 | 1998-11-19 | Chugai Seiyaku Kabushiki Kaisha | Indole derivatives and mono- and diazaindole derivatives |
| US5902726A (en) * | 1994-12-23 | 1999-05-11 | Glaxo Wellcome Inc. | Activators of the nuclear orphan receptor peroxisome proliferator-activated receptor gamma |
-
1999
- 1999-08-18 GB GBGB9919411.0A patent/GB9919411D0/en not_active Ceased
-
2000
- 2000-08-14 AU AU65834/00A patent/AU6583400A/en not_active Abandoned
- 2000-08-14 IL IL14782100A patent/IL147821A0/en unknown
- 2000-08-14 JP JP2001516533A patent/JP2003507327A/en active Pending
- 2000-08-14 CN CN00814336A patent/CN1379774A/en active Pending
- 2000-08-14 EP EP00953320A patent/EP1210343A2/en not_active Withdrawn
- 2000-08-14 MX MXPA02001598A patent/MXPA02001598A/en unknown
- 2000-08-14 NZ NZ517059A patent/NZ517059A/en unknown
- 2000-08-14 WO PCT/GB2000/003140 patent/WO2001012187A2/en active IP Right Grant
- 2000-08-14 BR BR0013368-0A patent/BR0013368A/en not_active IP Right Cessation
- 2000-08-14 KR KR1020027002019A patent/KR20020020817A/en not_active Application Discontinuation
- 2000-08-14 CA CA002380775A patent/CA2380775A1/en not_active Abandoned
-
2002
- 2002-01-24 ZA ZA200200669A patent/ZA200200669B/en unknown
- 2002-02-15 NO NO20020765A patent/NO20020765L/en not_active Application Discontinuation
Patent Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0179619A1 (en) * | 1984-10-19 | 1986-04-30 | Ici Americas Inc. | Heterocyclic amides |
| US4894386A (en) * | 1987-04-15 | 1990-01-16 | Ici Americas Inc. | Aliphatic carboxamides |
| US5902726A (en) * | 1994-12-23 | 1999-05-11 | Glaxo Wellcome Inc. | Activators of the nuclear orphan receptor peroxisome proliferator-activated receptor gamma |
| WO1997005114A1 (en) * | 1995-07-31 | 1997-02-13 | Laboratorios Menarini S.A. | Quinolone sulfonimides having leukotriene-antagonistic action |
| EP0780389A1 (en) * | 1995-12-22 | 1997-06-25 | Tobishi Pharmaceutical Co., Ltd. | Thiazolidinedione derivatives, process for their preparation and pharmaceutical compositions containing them |
| WO1998051667A1 (en) * | 1997-05-16 | 1998-11-19 | Chugai Seiyaku Kabushiki Kaisha | Indole derivatives and mono- and diazaindole derivatives |
Non-Patent Citations (4)
| Title |
|---|
| CHEMICAL ABSTRACTS, vol. 130, no. 9, 1 March 1999 (1999-03-01) Columbus, Ohio, US; abstract no. 110138m, HULME, CHRISTOPHER ET AL.: "Orally active indole N-oxide PDE4 inhibitors." XP002158920 & BIOORG. MED. CHEM. LETT., vol. 8, no. 21, - 1998 pages 3053-3058, -& DATABASE CHEMICAL ABSTRACTS [Online] CA 130:110138, XP002158921 * |
| R GRIERA ET AL.: "Synthesis and pharmacological evaluation of new cysLT1 receptor antagonists" EUROPEAN JOURNAL OF MEDICINAL CHEMISTRY.CHIMICA THERAPEUTICA., vol. 32, - 1997 pages 547-570, XP002158918 EDITIONS SCIENTIFIQUE ELSEVIER, PARIS., FR ISSN: 0223-5234 * |
| ROBERT T. JACOBS ET AL.: "Substituted 3-(phenylmethyl)-1H-indole-5-carboxamides and 1-(phenylmethyl)indole-6-carboxamides as potent, selective, orally active antagonists of the peptidoleukotrienes" JOURNAL OF MEDICINAL CHEMISTRY., vol. 36, no. 3, - 1993 pages 394-409, XP002158919 AMERICAN CHEMICAL SOCIETY. WASHINGTON., US ISSN: 0022-2623 * |
| ROBERT T. JACOBS ET AL.: "Synthesis, structure-activity relationships, and pharmacological evaluation of a series of fluorinated 3-benzyl-5-indolecarboxamides: identification of 4-((5-(((2R)-2-methyl-4,4,4,-trifluorobuty l)carbamoyl)-1-methylindol-3-yl)methyl)-3- methoxy-N-((2-methylphenyl)sulfonyl)benzam ide, ..." JOURNAL OF MEDICINAL CHEMISTRY., vol. 37, no. 9, - 1994 pages 1282-1297, XP002158917 AMERICAN CHEMICAL SOCIETY. WASHINGTON., US ISSN: 0022-2623 * |
Cited By (87)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2002044149A1 (en) * | 2000-11-28 | 2002-06-06 | F. Hoffmann-La Roche Ag | Indole and dihydroindole derivatives |
| EP1911462A2 (en) | 2001-01-26 | 2008-04-16 | Schering Corporation | Compositions comprising a sterol absorption inhibitor |
| US7074817B2 (en) | 2001-06-20 | 2006-07-11 | Wyeth | Substituted indole acid derivatives as inhibitors of plasminogen activator inhibitor-1 (PAI-1) |
| US7629377B2 (en) | 2001-06-20 | 2009-12-08 | Wyeth | Substituted naphthyl indole derivatives as inhibitors of plasminogen activator inhibitor type-1 (PAI-1) |
| US7291639B2 (en) | 2001-06-20 | 2007-11-06 | Wyeth | Aryloxy-acetic acid compounds useful as inhibitors of plasminogen activator inhibitor-1 (PAI-1) |
| US7368471B2 (en) | 2001-06-20 | 2008-05-06 | Wyeth | Substituted indole acid derivatives as inhibitors of plasminogen activator inhibitor-1 (PAI-1) |
| US7351730B2 (en) | 2001-06-20 | 2008-04-01 | Wyeth | Substituted naphthyl indole derivatives as inhibitors of plasminogen activator inhibitor type-1 (PAI-1) |
| WO2003035602A1 (en) * | 2001-10-25 | 2003-05-01 | Sankyo Company, Limited | Lipid modulators |
| US6984661B2 (en) | 2002-02-05 | 2006-01-10 | Eli Lilly And Company | Urea linker derivatives for use as PPAR modulators |
| WO2003066581A1 (en) * | 2002-02-05 | 2003-08-14 | Eli Lilly And Company | Urea linker derivatives for use as ppar modulators |
| US7355069B2 (en) | 2002-06-20 | 2008-04-08 | Astrazeneca Ab | Ortho-substituted benzoic acid derivatives for the treatment of insulin resistance |
| US7521461B2 (en) | 2002-06-20 | 2009-04-21 | Astrazeneca Ab | Benzoic acid derivatives as modulators of PPAR alpha and gamma |
| WO2004000295A1 (en) * | 2002-06-20 | 2003-12-31 | Astrazeneca Ab | Benzoic acid derivatives as modulators of ppar alpha and gamma |
| CN100400512C (en) * | 2002-06-20 | 2008-07-09 | 阿斯特拉曾尼卡有限公司 | Benzoic acid derivatives as modulators of PPAR alpha and gamma |
| US7160918B2 (en) | 2002-12-10 | 2007-01-09 | Hassan Mahmoud Elokdah | Substituted indole oxo-acetyl amino acetic acid derivatives as inhibitors of plasminogen activator inhibitor (PAI-1) |
| US7259182B2 (en) | 2002-12-10 | 2007-08-21 | Wyeth | Aryl, aryloxy, and aklyloxy substituted 1H-indol-3-yl glyoxylic acid derivatives as inhibitors of plasminogen activator inhibitor-1 (PAI-1) |
| US7566791B2 (en) | 2002-12-10 | 2009-07-28 | Wyeth | Substituted 3-carbonyl-1h-indol-1yl acetic acid derivatives as inhibitors of plasminogen activator inhibitor-1 (PAI-1) |
| US7101903B2 (en) | 2002-12-10 | 2006-09-05 | Wyeth | Substituted dihydropyrano indole-3,4-dione derivatives as inhibitiors of plasminogen activator inhibitor-1 (PAI-1) |
| US7348351B2 (en) | 2002-12-10 | 2008-03-25 | Wyeth | Substituted 3-alkyl and 3-arylalkyl 1H-indol-1yl acetic acid derivatives as inhibitors of plasminogen activator inhibitor-1 (PAI-1) |
| US7459478B2 (en) | 2002-12-10 | 2008-12-02 | Wyeth | Substituted dihydropyrano indole-3,4-dione derivatives as inhibitors of plasminogen activator inhibitor-1 (PAI-1) |
| US7078429B2 (en) | 2002-12-10 | 2006-07-18 | Wyeth | Substituted 3-carbonyl-1H-indol-1-yl acetic acid derivatives as inhibitors of plasminogen activator inhibitor-1 (PAI-1) |
| US7056943B2 (en) | 2002-12-10 | 2006-06-06 | Wyeth | Substituted indole oxo-acetyl amino acetic acid derivatives as inhibitors of plasminogen activator inhibitor-1 (PAI-1) |
| US7674818B2 (en) | 2002-12-10 | 2010-03-09 | Wyeth Llc | Aryl, aryloxy, alkyloxy substituted 1H-indol-3-yl glyoxylic acid derivatives as inhibitors of plasminogen activator inhibitor-1 (PAI-1) |
| US7476675B2 (en) | 2003-03-11 | 2009-01-13 | Roche Palo Alto Llc | Quinolinone derivatives and uses thereof |
| US6943169B2 (en) | 2003-03-11 | 2005-09-13 | Roche Palo Alto Llc | Quinolinone derivatives and uses thereof |
| US7129264B2 (en) | 2003-04-16 | 2006-10-31 | Bristol-Myers Squibb Company | Biarylmethyl indolines and indoles as antithromboembolic agents |
| WO2004096767A1 (en) * | 2003-04-25 | 2004-11-11 | H. Lundbeck A/S | Sustituted indoline and indole derivatives |
| US7897599B2 (en) | 2003-09-11 | 2011-03-01 | iTherX Pharmaceuticals Inc. | Cytokine inhibitors |
| US7749999B2 (en) | 2003-09-11 | 2010-07-06 | Itherx Pharmaceuticals, Inc. | Alpha-ketoamides and derivatives thereof |
| US7919617B2 (en) | 2003-09-11 | 2011-04-05 | iTherX Pharmaceuticals Inc. | Cytokine inhibitors |
| US7442805B2 (en) | 2003-09-25 | 2008-10-28 | Wyeth | Substituted sulfonamide-indoles |
| US7446201B2 (en) | 2003-09-25 | 2008-11-04 | Wyeth | Substituted heteroaryl benzofuran acids |
| US7351726B2 (en) | 2003-09-25 | 2008-04-01 | Wyeth | Substituted oxadiazolidinediones |
| US7803835B2 (en) | 2003-09-25 | 2010-09-28 | Wyeth Llc | Substituted acetic acid derivatives |
| US7582773B2 (en) | 2003-09-25 | 2009-09-01 | Wyeth | Substituted phenyl indoles |
| US7268159B2 (en) | 2003-09-25 | 2007-09-11 | Wyeth | Substituted indoles |
| US7163954B2 (en) | 2003-09-25 | 2007-01-16 | Wyeth | Substituted naphthyl benzothiophene acids |
| US7754747B2 (en) | 2004-08-23 | 2010-07-13 | Wyeth Llc | Oxazolo-naphthyl acids |
| EA013123B1 (en) * | 2005-04-13 | 2010-02-26 | Ньюрэксон, Инк. | Substituted indole compounds having nos inhibitory activity |
| WO2007063418A3 (en) * | 2005-04-13 | 2007-12-21 | Neuraxon Inc | Substituted indole compounds having nos inhibitory activity |
| US8586620B2 (en) | 2005-04-13 | 2013-11-19 | Neuraxon, Inc. | Substituted indole compounds having NOS inhibitory activity |
| AU2006321284B2 (en) * | 2005-04-13 | 2012-06-21 | Neuraxon, Inc. | Substituted indole compounds having NOS inhibitory activity |
| US7375219B2 (en) | 2005-04-13 | 2008-05-20 | Neuraxon, Inc. | Substituted indole compounds having NOS inhibitory activity |
| KR101463572B1 (en) * | 2005-04-13 | 2014-11-26 | 네우렉슨 인코포레이티드 | Substituted indole compounds having nos inhibitory activity |
| US7951940B2 (en) | 2005-04-13 | 2011-05-31 | Neuraxon, Inc. | Substituted indole compounds having NOS inhibitory activity |
| WO2007063418A2 (en) * | 2005-04-13 | 2007-06-07 | Neuraxon, Inc. | Substituted indole compounds having nos inhibitory activity |
| US7683091B2 (en) | 2005-08-17 | 2010-03-23 | Wyeth | Substituted indoles and methods of their use |
| US8686002B2 (en) | 2005-08-21 | 2014-04-01 | AbbVie Deutschland GmbH & Co. KG | Heterocyclic compounds and their use as binding partners for 5-HT5 receptors |
| US7989447B2 (en) | 2006-04-13 | 2011-08-02 | Neuraxon, Inc. | 1,5 and 3,6-substituted indole compounds having NOS inhibitory activity |
| US7928238B2 (en) | 2006-05-11 | 2011-04-19 | Janssen Pharmaceutica Nv | 1,2,3,4-tetrahydro-quinoline derivatives as CETP inhibitors |
| US8012963B2 (en) | 2006-05-11 | 2011-09-06 | Janssen Pharmaceutica N.V. | 3,4-dihydro-2H-benzo[1,4]oxazine and thiazine derivatives as CETP inhibitors |
| US7749995B2 (en) | 2006-05-11 | 2010-07-06 | Janssen Pharmaceutica Nv | 3,4-dihydro-2h-benzo[1,4]oxazine and thiazine derivatives as CETP inhibitors |
| WO2008020302A2 (en) * | 2006-08-17 | 2008-02-21 | Pfizer Products Inc. | Heteroaromatic quinoline-based compounds as phosphodiesterase (pde) inhibitors |
| WO2008020302A3 (en) * | 2006-08-17 | 2008-04-17 | Pfizer Prod Inc | Heteroaromatic quinoline-based compounds as phosphodiesterase (pde) inhibitors |
| WO2008109336A1 (en) | 2007-03-01 | 2008-09-12 | Janssen Pharmaceutica N.V. | Tetrahydroisoquinoline compounds as modulators of the histamine h3 receptor |
| US8093265B2 (en) | 2007-03-09 | 2012-01-10 | Renovis, Inc. | Bicycloheteroaryl compounds as P2X7 modulators and uses thereof |
| CN101877966A (en) * | 2007-07-02 | 2010-11-03 | 葛兰素史密丝克莱恩有限责任公司 | Farnesoid X receptor agonists |
| WO2009005998A1 (en) * | 2007-07-02 | 2009-01-08 | Smithkline Beecham Corporation | Farnesoid x receptor agonists |
| FR2921366A1 (en) * | 2007-09-26 | 2009-03-27 | Servier Lab | New heterocyclic compounds are aldose reductase inhibitors useful to treat e.g. hyperglycemia, dyslipidemia, non-insulin-dependant type II diabetes, syndrome X, coronary artery diseases, glomerulonephritis and psoriasis |
| US8673909B2 (en) | 2007-11-16 | 2014-03-18 | Neuraxon, Inc. | Indole compounds and methods for treating visceral pain |
| WO2009100294A3 (en) * | 2008-02-07 | 2009-11-12 | Abbott Laboratories | Amide derivatives as positive allosteric modulators and methods of use thereof |
| US8536221B2 (en) | 2008-02-07 | 2013-09-17 | Abbvie Inc. | Amide derivatives as positive allosteric modulators and methods of use thereof |
| WO2012027331A1 (en) | 2010-08-27 | 2012-03-01 | Ironwood Pharmaceuticals, Inc. | Compositions and methods for treating or preventing metabolic syndrome and related diseases and disorders |
| EP3023102A1 (en) | 2010-11-08 | 2016-05-25 | Albireo AB | Ibat inhibitors for the treatment of liver diseases |
| EP3777864A1 (en) | 2010-11-08 | 2021-02-17 | Albireo AB | Ibat inhibitors for the treatment of liver diseases |
| WO2012064266A1 (en) | 2010-11-08 | 2012-05-18 | Albireo Ab | Ibat inhibitors for the treatment of liver diseases |
| WO2012064267A1 (en) | 2010-11-08 | 2012-05-18 | Albireo Ab | A pharmaceutical combination comprising an ibat inhibitor and a bile acid binder |
| US9051265B2 (en) | 2011-06-06 | 2015-06-09 | The Scripps Research Institute | N-benzylindole modulators of PPARG |
| WO2012170561A1 (en) * | 2011-06-06 | 2012-12-13 | The Scripps Research Institute (T.S.R.I.) | N-benzylindole modulators of pparg |
| US20120309769A1 (en) * | 2011-06-06 | 2012-12-06 | Scripps Research Institute, The | N-benzylindole modulators of pparg |
| US8957093B2 (en) | 2011-06-06 | 2015-02-17 | The Scripps Research Institute | N-biphenylmethylindole modulators of PPARG |
| EP2736330A4 (en) * | 2011-07-29 | 2015-05-27 | Tempero Pharmaceuticals Inc | Compounds and methods |
| US9309227B2 (en) | 2011-11-22 | 2016-04-12 | The Scripps Research Institute | N-biphenylmethylbenzimidazole modulators of PPARG |
| US10988474B2 (en) | 2011-12-21 | 2021-04-27 | Allergan, Inc. | Compounds acting at multiple prostaglandin receptors giving a general anti-inflammatory response |
| WO2013096496A3 (en) * | 2011-12-21 | 2013-08-15 | Allergan, Inc. | Compounds acting at multiple prostaglandin receptors giving a general anti-inflammatory response |
| EP3424904A1 (en) * | 2011-12-21 | 2019-01-09 | Allergan, Inc. | Compounds acting at multiple prostaglandin receptors giving a general anti-inflammatory response |
| US9567328B2 (en) | 2011-12-21 | 2017-02-14 | Allergan, Inc. | Compounds acting at multiple prostaglandin receptors giving a general anti-inflammatory response |
| US20130184463A1 (en) * | 2011-12-21 | 2013-07-18 | Allergan, Inc. | Compounds acting at multiple prostaglandin receptors giving a general anti-inflammatory response |
| US10392382B2 (en) | 2011-12-21 | 2019-08-27 | Allergan, Inc. | Compounds acting at multiple prostaglandin receptors giving a general anti-inflammatory response |
| US9321750B2 (en) | 2012-04-20 | 2016-04-26 | Innov17 Llc | ROR modulators and their uses |
| EP2844247A4 (en) * | 2012-04-20 | 2015-11-25 | Anderson Gaweco | Ror modulators and their uses |
| US9708268B2 (en) | 2012-04-30 | 2017-07-18 | Innov17 Llc | ROR modulators and their uses |
| US10016394B2 (en) | 2014-04-16 | 2018-07-10 | The Scripps Research Institute | PPARG modulators for treatment of osteoporosis |
| WO2018214959A1 (en) * | 2017-05-26 | 2018-11-29 | 南京明德新药研发股份有限公司 | Lactam compound as fxr receptor agonist |
| US11339147B2 (en) | 2017-05-26 | 2022-05-24 | Cspc Zhongqi Pharmaceutical Technology (Shijiazhuang) Co., Ltd. | Lactam compound as FXR receptor agonist |
| RU2776052C2 (en) * | 2017-05-26 | 2022-07-12 | СиЭсПиСи ЧЖУНЦИ ФАРМАСЬЮТИКАЛ ТЕКНОЛОДЖИ (ШИЦЗЯЧЖУАН) КО., ЛТД. | Lactam compound as fxr receptor agonist |
| WO2020045982A1 (en) * | 2018-08-29 | 2020-03-05 | 숙명여자대학교산학협력단 | SUBSTITUTED INDOLE DERIVATIVE, PREPARATION METHOD FOR SAME, AND PHARMACEUTICAL COMPOSITION COMPRISING SAME AS EFFECTIVE COMPONENT FOR PREVENTING OR TREATING DISEASES ASSOCIATED WITH PPARα, PPARγ, AND PPARδ |
Also Published As
| Publication number | Publication date |
|---|---|
| NO20020765D0 (en) | 2002-02-15 |
| MXPA02001598A (en) | 2002-07-02 |
| AU6583400A (en) | 2001-03-13 |
| EP1210343A2 (en) | 2002-06-05 |
| KR20020020817A (en) | 2002-03-15 |
| WO2001012187A3 (en) | 2001-06-07 |
| CN1379774A (en) | 2002-11-13 |
| NZ517059A (en) | 2004-05-28 |
| IL147821A0 (en) | 2002-08-14 |
| BR0013368A (en) | 2002-05-07 |
| ZA200200669B (en) | 2003-06-25 |
| JP2003507327A (en) | 2003-02-25 |
| CA2380775A1 (en) | 2001-02-22 |
| NO20020765L (en) | 2002-04-17 |
| GB9919411D0 (en) | 1999-10-20 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP1210343A2 (en) | Benzoic acid derivatives and their use as ppar receptor agonists | |
| US6787556B1 (en) | Benzoic acid derivatives for the treatment of diabetes mellitus | |
| CN105683157A (en) | Sulfonamides as modulators of sodium channels | |
| EP0779887B1 (en) | Novel benzimidazole derivatives having cgmp-phosphodiesterase inhibiting activity | |
| JPH0696581B2 (en) | A new sulfenamide | |
| JP4662979B2 (en) | Novel β-agonist, its production method and its use as a drug | |
| KR20080114711A (en) | 17beta; hsd type 5 inhibitor | |
| TWI510240B (en) | Glucocorticoid receptor agonist consisting of derivatives of 2,2,4-trimethyl-6-phenyl-1,2-dihydroquinoline with substituted oxy group | |
| EP1870099A1 (en) | Protective agent for retinal neuronal cell comprising indazole derivative as active ingredient | |
| US20130040988A1 (en) | 3-amino-pyridine derivatives for the treatment of metabolic disorders | |
| JP4007743B2 (en) | Angiogenesis inhibitor | |
| JP4817577B2 (en) | Tyrosine derivatives with anti-leukotriene activity | |
| KR102459766B1 (en) | Polymorph form of 4-{[4-({[4-(2,2,2-trifluoroethoxy)-1,2-benzisoxazol-3-yl]oxy}methyl)piperidin-1-yl]methyl}-tetrahydro-2h-pyran-4-carboxylic acid | |
| CN102946882A (en) | Phenylalanine derivatives and their use as non-peptide glp-1 receptor modulators | |
| KR20100046107A (en) | Benzimidazole derivative | |
| JPH0633253B2 (en) | Novel benzimidazole derivative | |
| TWI297008B (en) | Novel tetrahydroisoquinoline derivates and pharmaceutical use thereof | |
| JP2002510293A (en) | Novel benzimidazole derivatives as anti-ulcer agents, methods for their preparation, and pharmaceutical compositions containing them | |
| KR940000785B1 (en) | Process for the preparation of carbostyril derivatives and salts thereof | |
| KR100464526B1 (en) | Sodium-hydrogen exchanger type 1 inhibitor crystals | |
| JP2821674B2 (en) | Benzimidazole derivatives | |
| JPH06345731A (en) | 2-(2-(indol-3-ly)ethylamino)-1-phenylethanol derivative | |
| WO2020114457A1 (en) | N-acyl sulfonamide salt fbpase inhibitor, preparation method therefor, pharmaceutical composition, and uses thereof | |
| JPH0770083A (en) | Antihypertensive agent containing imidazole derivative as active ingredient | |
| KR20070032264A (en) | Compounds having crth2 antagonist activity |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A2 Designated state(s): AE AG AL AM AT AU AZ BA BB BG BR BY CA CH CN CR CU CZ DE DK DM DZ EE ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MA MD MG MK MN MW MX NO NZ PL PT RO RU SD SE SG SI SK SL TJ TM TR TT TZ UA UG US UZ VN YU ZA ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A2 Designated state(s): GH GM KE LS MW MZ SD SL SZ TZ UG ZW AM AZ BY KG KZ MD RU TJ TM AT BE CH CY DE DK ES FI FR GB GR IE IT LU MC NL PT SE BF BJ CF CG CI CM GA GN GW ML MR NE SN TD TG |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| AK | Designated states |
Kind code of ref document: A3 Designated state(s): AE AG AL AM AT AU AZ BA BB BG BR BY CA CH CN CR CU CZ DE DK DM DZ EE ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MA MD MG MK MN MW MX NO NZ PL PT RO RU SD SE SG SI SK SL TJ TM TR TT TZ UA UG US UZ VN YU ZA ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A3 Designated state(s): GH GM KE LS MW MZ SD SL SZ TZ UG ZW AM AZ BY KG KZ MD RU TJ TM AT BE CH CY DE DK ES FI FR GB GR IE IT LU MC NL PT SE BF BJ CF CG CI CM GA GN GW ML MR NE SN TD TG |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2002/00669 Country of ref document: ZA Ref document number: 147821 Country of ref document: IL Ref document number: 200200669 Country of ref document: ZA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 65834/00 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2000953320 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2380775 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 517059 Country of ref document: NZ |
|
| WWE | Wipo information: entry into national phase |
Ref document number: PA/a/2002/001598 Country of ref document: MX |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1020027002019 Country of ref document: KR |
|
| WWP | Wipo information: published in national office |
Ref document number: 1020027002019 Country of ref document: KR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 008143366 Country of ref document: CN |
|
| WWP | Wipo information: published in national office |
Ref document number: 2000953320 Country of ref document: EP |
|
| REG | Reference to national code |
Ref country code: DE Ref legal event code: 8642 |
|
| WWP | Wipo information: published in national office |
Ref document number: 517059 Country of ref document: NZ |
|
| WWW | Wipo information: withdrawn in national office |
Ref document number: 2000953320 Country of ref document: EP |
|
| WWG | Wipo information: grant in national office |
Ref document number: 517059 Country of ref document: NZ |