US20040254635A1 - Expandable medical device for delivery of beneficial agent - Google Patents
Expandable medical device for delivery of beneficial agent Download PDFInfo
- Publication number
- US20040254635A1 US20040254635A1 US10/857,201 US85720104A US2004254635A1 US 20040254635 A1 US20040254635 A1 US 20040254635A1 US 85720104 A US85720104 A US 85720104A US 2004254635 A1 US2004254635 A1 US 2004254635A1
- Authority
- US
- United States
- Prior art keywords
- drug
- agent
- beneficial agent
- drugs
- stent
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Abandoned
Links
Images












Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61L—METHODS OR APPARATUS FOR STERILISING MATERIALS OR OBJECTS IN GENERAL; DISINFECTION, STERILISATION OR DEODORISATION OF AIR; CHEMICAL ASPECTS OF BANDAGES, DRESSINGS, ABSORBENT PADS OR SURGICAL ARTICLES; MATERIALS FOR BANDAGES, DRESSINGS, ABSORBENT PADS OR SURGICAL ARTICLES
- A61L31/00—Materials for other surgical articles, e.g. stents, stent-grafts, shunts, surgical drapes, guide wires, materials for adhesion prevention, occluding devices, surgical gloves, tissue fixation devices
- A61L31/08—Materials for coatings
- A61L31/10—Macromolecular materials
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61F—FILTERS IMPLANTABLE INTO BLOOD VESSELS; PROSTHESES; DEVICES PROVIDING PATENCY TO, OR PREVENTING COLLAPSING OF, TUBULAR STRUCTURES OF THE BODY, e.g. STENTS; ORTHOPAEDIC, NURSING OR CONTRACEPTIVE DEVICES; FOMENTATION; TREATMENT OR PROTECTION OF EYES OR EARS; BANDAGES, DRESSINGS OR ABSORBENT PADS; FIRST-AID KITS
- A61F2/00—Filters implantable into blood vessels; Prostheses, i.e. artificial substitutes or replacements for parts of the body; Appliances for connecting them with the body; Devices providing patency to, or preventing collapsing of, tubular structures of the body, e.g. stents
- A61F2/82—Devices providing patency to, or preventing collapsing of, tubular structures of the body, e.g. stents
- A61F2/856—Single tubular stent with a side portal passage
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61F—FILTERS IMPLANTABLE INTO BLOOD VESSELS; PROSTHESES; DEVICES PROVIDING PATENCY TO, OR PREVENTING COLLAPSING OF, TUBULAR STRUCTURES OF THE BODY, e.g. STENTS; ORTHOPAEDIC, NURSING OR CONTRACEPTIVE DEVICES; FOMENTATION; TREATMENT OR PROTECTION OF EYES OR EARS; BANDAGES, DRESSINGS OR ABSORBENT PADS; FIRST-AID KITS
- A61F2/00—Filters implantable into blood vessels; Prostheses, i.e. artificial substitutes or replacements for parts of the body; Appliances for connecting them with the body; Devices providing patency to, or preventing collapsing of, tubular structures of the body, e.g. stents
- A61F2/82—Devices providing patency to, or preventing collapsing of, tubular structures of the body, e.g. stents
- A61F2/86—Stents in a form characterised by the wire-like elements; Stents in the form characterised by a net-like or mesh-like structure
- A61F2/90—Stents in a form characterised by the wire-like elements; Stents in the form characterised by a net-like or mesh-like structure characterised by a net-like or mesh-like structure
- A61F2/91—Stents in a form characterised by the wire-like elements; Stents in the form characterised by a net-like or mesh-like structure characterised by a net-like or mesh-like structure made from perforated sheet material or tubes, e.g. perforated by laser cuts or etched holes
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61F—FILTERS IMPLANTABLE INTO BLOOD VESSELS; PROSTHESES; DEVICES PROVIDING PATENCY TO, OR PREVENTING COLLAPSING OF, TUBULAR STRUCTURES OF THE BODY, e.g. STENTS; ORTHOPAEDIC, NURSING OR CONTRACEPTIVE DEVICES; FOMENTATION; TREATMENT OR PROTECTION OF EYES OR EARS; BANDAGES, DRESSINGS OR ABSORBENT PADS; FIRST-AID KITS
- A61F2/00—Filters implantable into blood vessels; Prostheses, i.e. artificial substitutes or replacements for parts of the body; Appliances for connecting them with the body; Devices providing patency to, or preventing collapsing of, tubular structures of the body, e.g. stents
- A61F2/82—Devices providing patency to, or preventing collapsing of, tubular structures of the body, e.g. stents
- A61F2/86—Stents in a form characterised by the wire-like elements; Stents in the form characterised by a net-like or mesh-like structure
- A61F2/90—Stents in a form characterised by the wire-like elements; Stents in the form characterised by a net-like or mesh-like structure characterised by a net-like or mesh-like structure
- A61F2/91—Stents in a form characterised by the wire-like elements; Stents in the form characterised by a net-like or mesh-like structure characterised by a net-like or mesh-like structure made from perforated sheet material or tubes, e.g. perforated by laser cuts or etched holes
- A61F2/915—Stents in a form characterised by the wire-like elements; Stents in the form characterised by a net-like or mesh-like structure characterised by a net-like or mesh-like structure made from perforated sheet material or tubes, e.g. perforated by laser cuts or etched holes with bands having a meander structure, adjacent bands being connected to each other
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61L—METHODS OR APPARATUS FOR STERILISING MATERIALS OR OBJECTS IN GENERAL; DISINFECTION, STERILISATION OR DEODORISATION OF AIR; CHEMICAL ASPECTS OF BANDAGES, DRESSINGS, ABSORBENT PADS OR SURGICAL ARTICLES; MATERIALS FOR BANDAGES, DRESSINGS, ABSORBENT PADS OR SURGICAL ARTICLES
- A61L31/00—Materials for other surgical articles, e.g. stents, stent-grafts, shunts, surgical drapes, guide wires, materials for adhesion prevention, occluding devices, surgical gloves, tissue fixation devices
- A61L31/14—Materials characterised by their function or physical properties, e.g. injectable or lubricating compositions, shape-memory materials, surface modified materials
- A61L31/146—Porous materials, e.g. foams or sponges
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61L—METHODS OR APPARATUS FOR STERILISING MATERIALS OR OBJECTS IN GENERAL; DISINFECTION, STERILISATION OR DEODORISATION OF AIR; CHEMICAL ASPECTS OF BANDAGES, DRESSINGS, ABSORBENT PADS OR SURGICAL ARTICLES; MATERIALS FOR BANDAGES, DRESSINGS, ABSORBENT PADS OR SURGICAL ARTICLES
- A61L31/00—Materials for other surgical articles, e.g. stents, stent-grafts, shunts, surgical drapes, guide wires, materials for adhesion prevention, occluding devices, surgical gloves, tissue fixation devices
- A61L31/14—Materials characterised by their function or physical properties, e.g. injectable or lubricating compositions, shape-memory materials, surface modified materials
- A61L31/148—Materials at least partially resorbable by the body
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61L—METHODS OR APPARATUS FOR STERILISING MATERIALS OR OBJECTS IN GENERAL; DISINFECTION, STERILISATION OR DEODORISATION OF AIR; CHEMICAL ASPECTS OF BANDAGES, DRESSINGS, ABSORBENT PADS OR SURGICAL ARTICLES; MATERIALS FOR BANDAGES, DRESSINGS, ABSORBENT PADS OR SURGICAL ARTICLES
- A61L31/00—Materials for other surgical articles, e.g. stents, stent-grafts, shunts, surgical drapes, guide wires, materials for adhesion prevention, occluding devices, surgical gloves, tissue fixation devices
- A61L31/14—Materials characterised by their function or physical properties, e.g. injectable or lubricating compositions, shape-memory materials, surface modified materials
- A61L31/16—Biologically active materials, e.g. therapeutic substances
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61F—FILTERS IMPLANTABLE INTO BLOOD VESSELS; PROSTHESES; DEVICES PROVIDING PATENCY TO, OR PREVENTING COLLAPSING OF, TUBULAR STRUCTURES OF THE BODY, e.g. STENTS; ORTHOPAEDIC, NURSING OR CONTRACEPTIVE DEVICES; FOMENTATION; TREATMENT OR PROTECTION OF EYES OR EARS; BANDAGES, DRESSINGS OR ABSORBENT PADS; FIRST-AID KITS
- A61F2/00—Filters implantable into blood vessels; Prostheses, i.e. artificial substitutes or replacements for parts of the body; Appliances for connecting them with the body; Devices providing patency to, or preventing collapsing of, tubular structures of the body, e.g. stents
- A61F2/82—Devices providing patency to, or preventing collapsing of, tubular structures of the body, e.g. stents
- A61F2002/821—Ostial stents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61F—FILTERS IMPLANTABLE INTO BLOOD VESSELS; PROSTHESES; DEVICES PROVIDING PATENCY TO, OR PREVENTING COLLAPSING OF, TUBULAR STRUCTURES OF THE BODY, e.g. STENTS; ORTHOPAEDIC, NURSING OR CONTRACEPTIVE DEVICES; FOMENTATION; TREATMENT OR PROTECTION OF EYES OR EARS; BANDAGES, DRESSINGS OR ABSORBENT PADS; FIRST-AID KITS
- A61F2/00—Filters implantable into blood vessels; Prostheses, i.e. artificial substitutes or replacements for parts of the body; Appliances for connecting them with the body; Devices providing patency to, or preventing collapsing of, tubular structures of the body, e.g. stents
- A61F2/82—Devices providing patency to, or preventing collapsing of, tubular structures of the body, e.g. stents
- A61F2/86—Stents in a form characterised by the wire-like elements; Stents in the form characterised by a net-like or mesh-like structure
- A61F2/90—Stents in a form characterised by the wire-like elements; Stents in the form characterised by a net-like or mesh-like structure characterised by a net-like or mesh-like structure
- A61F2/91—Stents in a form characterised by the wire-like elements; Stents in the form characterised by a net-like or mesh-like structure characterised by a net-like or mesh-like structure made from perforated sheet material or tubes, e.g. perforated by laser cuts or etched holes
- A61F2/915—Stents in a form characterised by the wire-like elements; Stents in the form characterised by a net-like or mesh-like structure characterised by a net-like or mesh-like structure made from perforated sheet material or tubes, e.g. perforated by laser cuts or etched holes with bands having a meander structure, adjacent bands being connected to each other
- A61F2002/91508—Stents in a form characterised by the wire-like elements; Stents in the form characterised by a net-like or mesh-like structure characterised by a net-like or mesh-like structure made from perforated sheet material or tubes, e.g. perforated by laser cuts or etched holes with bands having a meander structure, adjacent bands being connected to each other the meander having a difference in amplitude along the band
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61F—FILTERS IMPLANTABLE INTO BLOOD VESSELS; PROSTHESES; DEVICES PROVIDING PATENCY TO, OR PREVENTING COLLAPSING OF, TUBULAR STRUCTURES OF THE BODY, e.g. STENTS; ORTHOPAEDIC, NURSING OR CONTRACEPTIVE DEVICES; FOMENTATION; TREATMENT OR PROTECTION OF EYES OR EARS; BANDAGES, DRESSINGS OR ABSORBENT PADS; FIRST-AID KITS
- A61F2/00—Filters implantable into blood vessels; Prostheses, i.e. artificial substitutes or replacements for parts of the body; Appliances for connecting them with the body; Devices providing patency to, or preventing collapsing of, tubular structures of the body, e.g. stents
- A61F2/82—Devices providing patency to, or preventing collapsing of, tubular structures of the body, e.g. stents
- A61F2/86—Stents in a form characterised by the wire-like elements; Stents in the form characterised by a net-like or mesh-like structure
- A61F2/90—Stents in a form characterised by the wire-like elements; Stents in the form characterised by a net-like or mesh-like structure characterised by a net-like or mesh-like structure
- A61F2/91—Stents in a form characterised by the wire-like elements; Stents in the form characterised by a net-like or mesh-like structure characterised by a net-like or mesh-like structure made from perforated sheet material or tubes, e.g. perforated by laser cuts or etched holes
- A61F2/915—Stents in a form characterised by the wire-like elements; Stents in the form characterised by a net-like or mesh-like structure characterised by a net-like or mesh-like structure made from perforated sheet material or tubes, e.g. perforated by laser cuts or etched holes with bands having a meander structure, adjacent bands being connected to each other
- A61F2002/91516—Stents in a form characterised by the wire-like elements; Stents in the form characterised by a net-like or mesh-like structure characterised by a net-like or mesh-like structure made from perforated sheet material or tubes, e.g. perforated by laser cuts or etched holes with bands having a meander structure, adjacent bands being connected to each other the meander having a change in frequency along the band
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61F—FILTERS IMPLANTABLE INTO BLOOD VESSELS; PROSTHESES; DEVICES PROVIDING PATENCY TO, OR PREVENTING COLLAPSING OF, TUBULAR STRUCTURES OF THE BODY, e.g. STENTS; ORTHOPAEDIC, NURSING OR CONTRACEPTIVE DEVICES; FOMENTATION; TREATMENT OR PROTECTION OF EYES OR EARS; BANDAGES, DRESSINGS OR ABSORBENT PADS; FIRST-AID KITS
- A61F2/00—Filters implantable into blood vessels; Prostheses, i.e. artificial substitutes or replacements for parts of the body; Appliances for connecting them with the body; Devices providing patency to, or preventing collapsing of, tubular structures of the body, e.g. stents
- A61F2/82—Devices providing patency to, or preventing collapsing of, tubular structures of the body, e.g. stents
- A61F2/86—Stents in a form characterised by the wire-like elements; Stents in the form characterised by a net-like or mesh-like structure
- A61F2/90—Stents in a form characterised by the wire-like elements; Stents in the form characterised by a net-like or mesh-like structure characterised by a net-like or mesh-like structure
- A61F2/91—Stents in a form characterised by the wire-like elements; Stents in the form characterised by a net-like or mesh-like structure characterised by a net-like or mesh-like structure made from perforated sheet material or tubes, e.g. perforated by laser cuts or etched holes
- A61F2/915—Stents in a form characterised by the wire-like elements; Stents in the form characterised by a net-like or mesh-like structure characterised by a net-like or mesh-like structure made from perforated sheet material or tubes, e.g. perforated by laser cuts or etched holes with bands having a meander structure, adjacent bands being connected to each other
- A61F2002/91525—Stents in a form characterised by the wire-like elements; Stents in the form characterised by a net-like or mesh-like structure characterised by a net-like or mesh-like structure made from perforated sheet material or tubes, e.g. perforated by laser cuts or etched holes with bands having a meander structure, adjacent bands being connected to each other within the whole structure different bands showing different meander characteristics, e.g. frequency or amplitude
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61F—FILTERS IMPLANTABLE INTO BLOOD VESSELS; PROSTHESES; DEVICES PROVIDING PATENCY TO, OR PREVENTING COLLAPSING OF, TUBULAR STRUCTURES OF THE BODY, e.g. STENTS; ORTHOPAEDIC, NURSING OR CONTRACEPTIVE DEVICES; FOMENTATION; TREATMENT OR PROTECTION OF EYES OR EARS; BANDAGES, DRESSINGS OR ABSORBENT PADS; FIRST-AID KITS
- A61F2/00—Filters implantable into blood vessels; Prostheses, i.e. artificial substitutes or replacements for parts of the body; Appliances for connecting them with the body; Devices providing patency to, or preventing collapsing of, tubular structures of the body, e.g. stents
- A61F2/82—Devices providing patency to, or preventing collapsing of, tubular structures of the body, e.g. stents
- A61F2/86—Stents in a form characterised by the wire-like elements; Stents in the form characterised by a net-like or mesh-like structure
- A61F2/90—Stents in a form characterised by the wire-like elements; Stents in the form characterised by a net-like or mesh-like structure characterised by a net-like or mesh-like structure
- A61F2/91—Stents in a form characterised by the wire-like elements; Stents in the form characterised by a net-like or mesh-like structure characterised by a net-like or mesh-like structure made from perforated sheet material or tubes, e.g. perforated by laser cuts or etched holes
- A61F2/915—Stents in a form characterised by the wire-like elements; Stents in the form characterised by a net-like or mesh-like structure characterised by a net-like or mesh-like structure made from perforated sheet material or tubes, e.g. perforated by laser cuts or etched holes with bands having a meander structure, adjacent bands being connected to each other
- A61F2002/91533—Stents in a form characterised by the wire-like elements; Stents in the form characterised by a net-like or mesh-like structure characterised by a net-like or mesh-like structure made from perforated sheet material or tubes, e.g. perforated by laser cuts or etched holes with bands having a meander structure, adjacent bands being connected to each other characterised by the phase between adjacent bands
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61F—FILTERS IMPLANTABLE INTO BLOOD VESSELS; PROSTHESES; DEVICES PROVIDING PATENCY TO, OR PREVENTING COLLAPSING OF, TUBULAR STRUCTURES OF THE BODY, e.g. STENTS; ORTHOPAEDIC, NURSING OR CONTRACEPTIVE DEVICES; FOMENTATION; TREATMENT OR PROTECTION OF EYES OR EARS; BANDAGES, DRESSINGS OR ABSORBENT PADS; FIRST-AID KITS
- A61F2/00—Filters implantable into blood vessels; Prostheses, i.e. artificial substitutes or replacements for parts of the body; Appliances for connecting them with the body; Devices providing patency to, or preventing collapsing of, tubular structures of the body, e.g. stents
- A61F2/82—Devices providing patency to, or preventing collapsing of, tubular structures of the body, e.g. stents
- A61F2/86—Stents in a form characterised by the wire-like elements; Stents in the form characterised by a net-like or mesh-like structure
- A61F2/90—Stents in a form characterised by the wire-like elements; Stents in the form characterised by a net-like or mesh-like structure characterised by a net-like or mesh-like structure
- A61F2/91—Stents in a form characterised by the wire-like elements; Stents in the form characterised by a net-like or mesh-like structure characterised by a net-like or mesh-like structure made from perforated sheet material or tubes, e.g. perforated by laser cuts or etched holes
- A61F2/915—Stents in a form characterised by the wire-like elements; Stents in the form characterised by a net-like or mesh-like structure characterised by a net-like or mesh-like structure made from perforated sheet material or tubes, e.g. perforated by laser cuts or etched holes with bands having a meander structure, adjacent bands being connected to each other
- A61F2002/91533—Stents in a form characterised by the wire-like elements; Stents in the form characterised by a net-like or mesh-like structure characterised by a net-like or mesh-like structure made from perforated sheet material or tubes, e.g. perforated by laser cuts or etched holes with bands having a meander structure, adjacent bands being connected to each other characterised by the phase between adjacent bands
- A61F2002/91541—Adjacent bands are arranged out of phase
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61F—FILTERS IMPLANTABLE INTO BLOOD VESSELS; PROSTHESES; DEVICES PROVIDING PATENCY TO, OR PREVENTING COLLAPSING OF, TUBULAR STRUCTURES OF THE BODY, e.g. STENTS; ORTHOPAEDIC, NURSING OR CONTRACEPTIVE DEVICES; FOMENTATION; TREATMENT OR PROTECTION OF EYES OR EARS; BANDAGES, DRESSINGS OR ABSORBENT PADS; FIRST-AID KITS
- A61F2/00—Filters implantable into blood vessels; Prostheses, i.e. artificial substitutes or replacements for parts of the body; Appliances for connecting them with the body; Devices providing patency to, or preventing collapsing of, tubular structures of the body, e.g. stents
- A61F2/82—Devices providing patency to, or preventing collapsing of, tubular structures of the body, e.g. stents
- A61F2/86—Stents in a form characterised by the wire-like elements; Stents in the form characterised by a net-like or mesh-like structure
- A61F2/90—Stents in a form characterised by the wire-like elements; Stents in the form characterised by a net-like or mesh-like structure characterised by a net-like or mesh-like structure
- A61F2/91—Stents in a form characterised by the wire-like elements; Stents in the form characterised by a net-like or mesh-like structure characterised by a net-like or mesh-like structure made from perforated sheet material or tubes, e.g. perforated by laser cuts or etched holes
- A61F2/915—Stents in a form characterised by the wire-like elements; Stents in the form characterised by a net-like or mesh-like structure characterised by a net-like or mesh-like structure made from perforated sheet material or tubes, e.g. perforated by laser cuts or etched holes with bands having a meander structure, adjacent bands being connected to each other
- A61F2002/9155—Adjacent bands being connected to each other
- A61F2002/91558—Adjacent bands being connected to each other connected peak to peak
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61F—FILTERS IMPLANTABLE INTO BLOOD VESSELS; PROSTHESES; DEVICES PROVIDING PATENCY TO, OR PREVENTING COLLAPSING OF, TUBULAR STRUCTURES OF THE BODY, e.g. STENTS; ORTHOPAEDIC, NURSING OR CONTRACEPTIVE DEVICES; FOMENTATION; TREATMENT OR PROTECTION OF EYES OR EARS; BANDAGES, DRESSINGS OR ABSORBENT PADS; FIRST-AID KITS
- A61F2250/00—Special features of prostheses classified in groups A61F2/00 - A61F2/26 or A61F2/82 or A61F9/00 or A61F11/00 or subgroups thereof
- A61F2250/0014—Special features of prostheses classified in groups A61F2/00 - A61F2/26 or A61F2/82 or A61F9/00 or A61F11/00 or subgroups thereof having different values of a given property or geometrical feature, e.g. mechanical property or material property, at different locations within the same prosthesis
- A61F2250/0035—Special features of prostheses classified in groups A61F2/00 - A61F2/26 or A61F2/82 or A61F9/00 or A61F11/00 or subgroups thereof having different values of a given property or geometrical feature, e.g. mechanical property or material property, at different locations within the same prosthesis differing in release or diffusion time
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61F—FILTERS IMPLANTABLE INTO BLOOD VESSELS; PROSTHESES; DEVICES PROVIDING PATENCY TO, OR PREVENTING COLLAPSING OF, TUBULAR STRUCTURES OF THE BODY, e.g. STENTS; ORTHOPAEDIC, NURSING OR CONTRACEPTIVE DEVICES; FOMENTATION; TREATMENT OR PROTECTION OF EYES OR EARS; BANDAGES, DRESSINGS OR ABSORBENT PADS; FIRST-AID KITS
- A61F2250/00—Special features of prostheses classified in groups A61F2/00 - A61F2/26 or A61F2/82 or A61F9/00 or A61F11/00 or subgroups thereof
- A61F2250/0058—Additional features; Implant or prostheses properties not otherwise provided for
- A61F2250/0067—Means for introducing or releasing pharmaceutical products into the body
- A61F2250/0068—Means for introducing or releasing pharmaceutical products into the body the pharmaceutical product being in a reservoir
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61L—METHODS OR APPARATUS FOR STERILISING MATERIALS OR OBJECTS IN GENERAL; DISINFECTION, STERILISATION OR DEODORISATION OF AIR; CHEMICAL ASPECTS OF BANDAGES, DRESSINGS, ABSORBENT PADS OR SURGICAL ARTICLES; MATERIALS FOR BANDAGES, DRESSINGS, ABSORBENT PADS OR SURGICAL ARTICLES
- A61L2300/00—Biologically active materials used in bandages, wound dressings, absorbent pads or medical devices
- A61L2300/40—Biologically active materials used in bandages, wound dressings, absorbent pads or medical devices characterised by a specific therapeutic activity or mode of action
- A61L2300/416—Anti-neoplastic or anti-proliferative or anti-restenosis or anti-angiogenic agents, e.g. paclitaxel, sirolimus
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61L—METHODS OR APPARATUS FOR STERILISING MATERIALS OR OBJECTS IN GENERAL; DISINFECTION, STERILISATION OR DEODORISATION OF AIR; CHEMICAL ASPECTS OF BANDAGES, DRESSINGS, ABSORBENT PADS OR SURGICAL ARTICLES; MATERIALS FOR BANDAGES, DRESSINGS, ABSORBENT PADS OR SURGICAL ARTICLES
- A61L2300/00—Biologically active materials used in bandages, wound dressings, absorbent pads or medical devices
- A61L2300/60—Biologically active materials used in bandages, wound dressings, absorbent pads or medical devices characterised by a special physical form
- A61L2300/602—Type of release, e.g. controlled, sustained, slow
Definitions
- the present invention relates to tissue-supporting medical devices, and more particularly to expandable, non-removable devices that are implanted within a bodily lumen of a living animal or human to support the organ and maintain patency, and that can deliver a beneficial agent to the intervention site.
- Known stent designs include monofilament wire coil stents (U.S. Pat. No. 4,969,458); welded metal cages (U.S. Pat. Nos. 4,733,665 and 4,776,337); and, most prominently, thin-walled metal cylinders with axial slots formed around the circumference (U.S. Pat. Nos. 4,733,665; 4,739,762; and 4,776,337).
- Known construction materials for use in stents include polymers, organic fabrics and biocompatible metals, such as, stainless steel, gold, silver, tantalum, titanium, and shape memory alloys such as Nitinol.
- U.S. Pat. Nos. 4,733,665; 4,739,762; and 4,776,337 disclose expandable and deformable interluminal vascular grafts in the form of thin-walled tubular members with axial slots allowing the members to be expanded radially outwardly into contact with a body passageway. After insertion, the tubular members are mechanically expanded beyond their elastic limit and thus permanently fixed within the body.
- U.S. Pat. No. 5,545,210 discloses a thin-walled tubular stent geometrically similar to those discussed above, but constructed of a nickel-titanium shape memory alloy (“Nitinol”), which can be permanently fixed within the body without exceeding its elastic limit.
- each design the features that undergo permanent deformation during stent expansion are prismatic, i.e., the cross sections of these features remain constant or change very gradually along their entire active length.
- prismatic structures are ideally suited to providing large amounts of elastic deformation before permanent deformation commences, which in turn leads to sub-optimal device performance in important properties including stent expansion force, stent recoil, strut element stability, stent securement on delivery catheters, and radiopacity.
- U.S. Pat. No. 6,241,762 which is incorporated herein by reference in its entirety, discloses a non-prismatic stent design which remedies the above mentioned performance deficiencies of previous stents.
- preferred embodiments of this patent provide a stent with large, non-deforming strut and link elements, which can contain holes without compromising the mechanical properties of the strut or link elements, or the device as a whole. Further, these holes may serve as large, protected reservoirs for delivering various beneficial agents to the device implantation site.
- restenosis is a major complication that can arise following vascular interventions such as angioplasty and the implantation of stents.
- vascular interventions such as angioplasty and the implantation of stents.
- restenosis is a wound healing process that reduces the vessel lumen diameter by extracellular matrix deposition and vascular smooth muscle cell proliferation, and which may ultimately result in renarrowing or even reocclusion of the lumen.
- the overall restenosis rate is still reported in the range of 25% to 50% within six to twelve months after an angioplasty procedure. To treat this condition, additional revascularization procedures are frequently required, thereby increasing trauma and risk to the patient.
- Some of the techniques under development to address the problem of restenosis include irradiation of the injury site and the use of conventional stents to deliver a variety of beneficial or pharmaceutical agents to the wall of the traumatized vessel.
- a conventional stent is frequently surface-coated with a beneficial agent (often a drug-impregnated polymer) and implanted at the angioplasty site.
- a beneficial agent often a drug-impregnated polymer
- an external drug-impregnated polymer sheath is mounted over the stent and co-deployed in the vessel.
- Increasing the thickness of the surface coating has the beneficial effects of improving drug release kinetics including the ability to control drug release and to allow increased drug loading.
- the increased coating thickness results in increased overall thickness of the stent wall. This is undesirable for a number of reasons, including increased trauma to the vessel wall during implantation, reduced flow cross-section of the lumen after implantation, and increased vulnerability of the coating to mechanical failure or damage during expansion and implantation.
- Coating thickness is one of several factors that affect the release kinetics of the beneficial agent, and limitations on thickness thereby limit the range of release rates, durations, and the like that can be achieved.
- Another significant problem is that expansion of the stent may stress the overlying polymeric coating causing the coating to plastically deform or even to rupture, which may therefore effect drug release kinetics or have other untoward effects. Further, expansion of such a coated stent in an atherosclerotic blood vessel will place circumferential shear forces on the polymeric coating, which may cause the coating to separate from the underlying stent surface. Such separation may again have untoward effects including embolization of coating fragments causing vascular obstruction.
- a device for the controlled release of one or more drugs comprises an implantable stent, at least two reservoirs in the stent, and a release system contained in each of the at least two reservoirs, wherein the release system comprises one or more drugs for release.
- FIG. 1 is a perspective view of a tissue supporting device in accordance with a first preferred embodiment of the present invention
- FIG. 2 is an enlarged side view of a portion of the device of FIG. 1;
- FIG. 3 is an enlarged side view of a tissue supporting device in accordance with a further preferred embodiment of the present invention.
- FIG. 4 is an enlarged side view of a portion of the stent shown in FIG. 3;
- FIG. 5 is an enlarged cross section of an opening
- FIG. 6 is an enlarged cross section of an opening illustrating beneficial agent loaded into the opening
- FIG. 7 is an enlarged cross section of an opening illustrating a beneficial agent loaded into the opening and a thin coating of a beneficial agent
- FIG. 8 is an enlarged cross section of an opening illustrating a beneficial agent loaded into the opening and thin coatings of different beneficial agents on different surfaces of the device;
- FIG. 9 is an enlarged cross section of an opening illustrating a beneficial agent provided in a plurality of layers
- FIG. 10 is an enlarged cross section of an opening illustrating a beneficial agent and a barrier layer loaded into the opening in layers;
- FIG. 11A is an enlarged cross section of an opening illustrating a beneficial agent, a biodegradable carrier, and a barrier layer loaded into the opening in layers;
- FIG. 11B is a graph of the release kinetics of the device of FIG. 11A;
- FIG. 12 is an enlarged cross section of an opening illustrating different beneficial agents, carrier, and barrier layers loaded into the opening;
- FIG. 13 is an enlarged cross section of an opening illustrating a beneficial agent loaded into the opening in layers of different concentrations
- FIG. 14 is an enlarged cross section of an opening illustrating a beneficial agent loaded into the opening in layers of microspheres of different sizes
- FIG. 15A is an enlarged cross section of a tapered opening illustrating a beneficial agent loaded into the opening
- FIG. 15B is an enlarged cross section of the tapered opening of FIG. 15A with the beneficial agent partially degraded;
- FIG. 15C is a graph of the release kinetics of the device of FIGS. 15A and 15B;
- FIG. 16A is an enlarged cross section of an opening illustrating a beneficial agent loaded into the opening in a shape configured to achieve a desired agent delivery profile
- FIG. 16B is an enlarged cross section of the opening of FIG. 16A with the beneficial agent partially degraded
- FIG. 16C is a graph of the release kinetics of the device of FIGS. 16A and 16B;
- FIG. 17A is an enlarged cross section of an opening illustrating the beneficial agent loaded into the opening and a spherical shape
- FIG. 17B is a graph of the release kinetics of the device of FIG. 17A;
- FIG. 18A is an enlarged cross section of an opening illustrating a plurality of beneficial agent layers and a barrier layer with an opening for achieving a desired agent delivery profile
- FIG. 18B is an enlarged cross section of the opening of FIG. 18A with the agent layers beginning to degraded;
- FIG. 18C is an enlarged cross section of the opening of FIG. 18A with the agent layers further degraded;
- FIG. 19 is an enlarged cross section of an opening illustrating a plurality of cylindrical beneficial agent layers
- FIG. 20 is an isometric view of an expandable tissue supporting device with different beneficial agents in different holes
- FIG. 21 is an isometric view of an expandable tissue supporting device with different beneficial agents in alternating holes.
- FIG. 22 is an enlarged side view of a portion of an expandable tissue supporting device with beneficial agent openings in the bridging elements.
- the tissue supporting device 10 includes a plurality of cylindrical tubes 12 connected by S-shaped bridging elements 14 .
- the bridging elements 14 allow the tissue supporting device to bend axially when passing through the tortuous path of the vasculature to the deployment site and allow the device to bend when necessary to match the curvature of a vessel wall to be supported.
- Each of the cylindrical tubes 12 has a plurality of axial slots 16 extending from an end surface of the cylindrical tube toward an opposite end surface.
- a network of axial struts 18 and links 22 Formed between the slots 16 is a network of axial struts 18 and links 22 .
- the struts 18 and links 22 are provided with openings for receiving and delivering a beneficial agent.
- the beneficial agent is loaded into the openings in layers or other configurations which provide control over the temporal release kinetics of the agent.
- Each individual strut 18 is preferably linked to the rest of the structure through a pair of reduced sections 20 , one at each end, which act as stress/strain concentration features.
- the reduced sections 20 of the struts function as hinges in the cylindrical structure. Since the stress/strain concentration features are designed to operate into the plastic deformation range of generally ductile materials, they are referred to as ductile hinges 20 .
- the ductile hinges 20 are described in further detail in U.S. Pat. No. 6,241,762, which has been incorporated herein by reference.
- the width of any feature is defined as its dimension in the circumferential direction of the cylinder.
- the length of any feature is defined as its dimension in the axial direction of the cylinder.
- the thickness of any feature is defined as the wall thickness of the cylinder.
- the presence of the ductile hinges 20 allows all of the remaining features in the tissue supporting device to be increased in width or the circumferentially oriented component of their respective rectangular moments of inertia—thus greatly increasing the strength and rigidity of these features.
- the net result is that elastic, and then plastic deformation commence and propagate in the ductile hinges 20 before other structural elements of the device undergo any significant elastic deformation.
- the force required to expand the tissue supporting device 10 becomes a function of the geometry of the ductile hinges 20 , rather than the device structure as a whole, and arbitrarily small expansion forces can be specified by changing hinge geometry for virtually any material wall thickness.
- the ability to increase the width and thickness of the struts 18 and links 22 provides additional area and depth for the beneficial agent receiving openings.
- the ductile hinge 20 illustrated in FIGS. 1 and 2 is exemplary of a preferred structure that will function as a stress/strain concentrator. Many other stress/strain concentrator configurations may also be used as the ductile hinges in the present invention, as shown and described by way of example in U.S. Pat. No. 6,241,762.
- the geometric details of the stress/strain concentration features or ductile hinges 20 can be varied greatly to tailor the exact mechanical expansion properties to those required in a specific application.
- FIG. 1 which includes ductile hinges
- the beneficial agent may be contained in openings in stents having a variety of designs including the designs illustrated in U.S. Provisional Patent Application Ser. No. 60/314,360, filed on Aug. 20, 2001 and U.S. patent application Ser. No. 09/948,987, filed on Sep. 7, 2001 (Attorney Docket No. 032304-033), which are incorporated herein by reference.
- the present invention incorporating beneficial agent openings may also be used with other known stent designs.
- openings 24 are formed by laser drilling or any other means known to one skilled in the art at intervals along the neutral axis of the struts 18 .
- at least one and preferably a series of openings 26 are formed at selected locations in the links 22 .
- openings 24 and 26 are circular in nature and form cylindrical holes extending through the width of the tissue supporting device 10 . It should be apparent to one skilled in the art, however, that openings of any geometrical shape or configuration could of course be used without departing from the scope of the present invention. In addition, openings having a depth less than the thickness of the device may also be used.
- the behavior of the struts 18 in bending is analogous to the behavior of an I-beam or truss.
- the outer edge elements 32 of the struts 18 shown in FIG. 2, correspond to the I-beam flange and carry the tensile and compressive stresses, whereas the inner elements 34 of the struts 18 correspond to the web of an I-beam which carries the shear and helps to prevent buckling and wrinkling of the faces. Since most of the bending load is carried by the outer edge elements 32 of the struts 18 , a concentration of as much material as possible away from the neutral axis results in the most efficient sections for resisting strut flexure.
- openings 24 , 26 are also non-deforming.
- the openings 24 , 26 in the struts 18 may promote the healing of the intervention site by promoting regrowth of the endothelial cells.
- the cross section of the strut is effectively reduced without decreasing the strength and integrity of the strut, as described above.
- the overall distance across which endothelial cell regrowth must occur is also reduced to approximately 0.0025-0.0035 inches, which is approximately one-half of the thickness of a conventional stent. It is further believed that during insertion of the expandable medical device, cells from the endothelial layer may be scraped from the inner wall of the vessel by the openings 24 , 26 and remain therein after implantation. The presence of such endothelial cells would thus provide a basis for the healing of the vessel wall.
- the openings 24 , 26 are loaded with an agent, most preferably a beneficial agent, for delivery to the vessel wall which the tissue supporting device 10 is supporting.
- agent and “beneficial agent” as used herein are intended to have their broadest possible interpretation and are used to include any therapeutic agent or drug, as well as inactive agents such as barrier layers or carrier layers.
- drug and “therapeutic agent” are used interchangeably to refer to any therapeutically active substance that is delivered to a bodily conduit of a living being to produce a desired, usually beneficial, effect.
- the present invention is particularly well suited for the delivery of antiproliferatives (anti-restenosis agents) such as paclitaxel and rapamycin for example, and antithrombins such as heparin, for example.
- anti-infectives such as antibiotics and antiviral agents
- analgesics including fentanyl, sufentanil, buprenorphine and analgesic combinations
- anesthetics including fentanyl, sufentanil, buprenorphine and analgesic combinations
- anesthetics including fentanyl, sufentanil, buprenorphine and analgesic combinations
- anesthetics anorexics
- antiarthritics such as terbutaline
- anticonvulsants antidepressants
- antidiabetic agents antidiarrheals
- antihistamines anti-inflammatory agents
- antimigraine preparations such as scopolamine and ondansetron
- antinauseants such as scopolamine and ondansetron
- antinauseants such as scopolamine and ondansetron
- antinauseants such as scopolamine and ondansetron
- the beneficial agents used in the present invention include classical small molecular weight therapeutic agents commonly referred to as drugs including all classes of action as exemplified by, but not limited to: antiproliferatives, antithrombins, antiplatelet, antilipid, anti-inflammatory, angiogenic, anti-angiogenic, vitamins, ACE inhibitors, vasoactive substances, antimitotics, metello-proteinase inhibitors, NO donors, estradiols, anti-sclerosing agents, alone or in combination.
- drugs including all classes of action as exemplified by, but not limited to: antiproliferatives, antithrombins, antiplatelet, antilipid, anti-inflammatory, angiogenic, anti-angiogenic, vitamins, ACE inhibitors, vasoactive substances, antimitotics, metello-proteinase inhibitors, NO donors, estradiols, anti-sclerosing agents, alone or in combination.
- Beneficial agent also includes larger molecular weight substances with drug like effects on target tissue sometimes called biologic agents including but not limited to: peptides, lipids, protein drugs, enzymes, oligonucleotides, ribozymes, genetic material, prions, virus, bacteria, and eucaryotic cells such as endothelial cells, monocyte/macrophages or vascular smooth muscle cells to name but a few examples.
- biologic agents including but not limited to: peptides, lipids, protein drugs, enzymes, oligonucleotides, ribozymes, genetic material, prions, virus, bacteria, and eucaryotic cells such as endothelial cells, monocyte/macrophages or vascular smooth muscle cells to name but a few examples.
- the therapeutic agent may also be a pro-drug, which metabolizes into the desired drug when administered to a host.
- beneficial agents may include but not be limited to physical agents such as microcapsules, microspheres, microbubbles, liposomes, niosomes, radioactive isotopes, emulsions, dispersions, or agents activated by some other form of energy such as light or ultrasonic energy, or by other circulating molecules that can be systemically administered.
- FIGS. 1 and 2 can be further refined by using Finite Element Analysis and other techniques to optimize the deployment of the beneficial agent within the openings of the struts and links.
- the shape and location of the openings 24 , 26 can be modified to maximize the volume of the voids while preserving the relatively high strength and rigidity of the struts 18 with respect to the ductile hinges 20 .
- FIG. 3 illustrates a further preferred embodiment of the present invention, wherein like reference numerals have been used to indicate like components.
- the tissue supporting device 100 includes a plurality of cylindrical tubes 12 connected by S-shaped bridging elements 14 .
- Each of the cylindrical tubes 12 has a plurality of axial slots 16 extending from an end surface of the cylindrical tube toward an opposite end surface. Formed between the slots 16 is a network of axial struts 18 and links 22 .
- Each individual strut 18 is linked to the rest of the structure through a pair of ductile hinges 20 , one at each end, which act as stress/strain concentration features.
- Each of the ductile hinges 20 is formed between an arc surface 28 and a concave notch surface 29 .
- openings 24 ′, 26 ′ in both the struts 18 and links 22 are preferred, it should be clear to one skilled in the art that openings could be formed in only one of the struts and links.
- the openings 24 ′ in the struts 18 are generally rectangular whereas the openings 26 ′ in the links 22 are polygonal.
- openings of any geometrical shape or configuration could of course be used, and that the shape of openings 24 , 24 ′ may be the same or different from the shape of openings 26 , 26 ′, without departing from the scope of the present invention.
- the openings 24 ′, 26 ′ may be loaded with an agent, most preferably a beneficial agent, for delivery to the vessel in which the tissue support device 100 is deployed.
- the openings 24 ′, 26 ′ are preferably through openings, they may also be recesses extending only partially through the thickness of the struts and links.
- the large through-openings in the expandable device of the present invention form protected areas or receptors to facilitate the loading of such an agent either at the time of use or prior to use, and to protect the agent from abrasion and extrusion during delivery and implantation.
- the volume of beneficial agent that can be delivered using through openings is about 3 to 10 times greater than the volume of a 5 micron coating covering a stent with the same stent/vessel wall coverage ratio.
- This much larger beneficial agent capacity provides several advantages.
- the larger capacity can be used to deliver multi-drug combinations, each with independent release profiles, for improved efficacy.
- larger capacity can be used to provide larger quantities of less aggressive drugs and to achieve clinical efficacy without the undesirable side-effects of more potent drugs, such as retarded healing of the endothelial layer.
- FIG. 4 shows an enlarged view of one of the struts 18 of device 100 disposed between a pair of ductile hinges 20 having a plurality of openings 24 ′.
- FIG. 5 illustrates a cross section of one of the openings 24 ′ shown in FIG. 4.
- FIG. 6 illustrates the same cross section when a beneficial agent 36 has been loaded into the opening 24 ′ of the strut 18 .
- the entire exterior surface of the stent can be coated with a thin layer of a beneficial agent 38 , which may be the same as or different from the beneficial agent 36 , as schematically shown in FIG. 7.
- FIG. 8 Another variation of the present invention would coat the outwardly facing surfaces of the stent with a first beneficial agent 38 while coating the inwardly facing surfaces of the stent with a different beneficial agent 39 , as illustrated in FIG. 8.
- the inwardly facing surface of the stent would be defined as at least the surface of the stent which, after expansion, forms the inner passage of the vessel.
- the outwardly facing surface of the stent would be defined as at least the surface of the stent which, after expansion, is in contact with and directly supports the inner wall of the vessel.
- the beneficial agent 39 coated on the inner surfaces may be a barrier layer which prevents the beneficial agent 36 from passing into the lumen of the blood vessel and being washed away in the blood stream.
- FIG. 9 shows a cross section of an opening 24 in which one or more beneficial agents have been loaded into the opening 24 in discrete layers 50 .
- One method of creating such layers is to deliver a solution comprising beneficial agent, polymer carrier, and a solvent into the opening and evaporating the solvent to create a thin solid layer of beneficial agent in the carrier.
- Other methods of delivering the beneficial agent can also be used to create layers.
- a beneficial agent may be loaded into the openings alone if the agent is structurally viable without the need for a carrier. The process can then be repeated until each opening is partially or entirely filled.
- the total depth of the opening 24 is about 125 to about 140 microns, and the typical layer thickness would be about 2 to about 50 microns, preferably about 12 microns.
- Each typical layer is thus individually about twice as thick as the typical coating applied to surface-coated stents.
- the openings have an area of at least 5 ⁇ 10 ⁇ 6 square inches, and preferably at least 7 ⁇ 10 ⁇ 6 square inches.
- each layer is created independently, individual chemical compositions and pharmacokinetic properties can be imparted to each layer. Numerous useful arrangements of such layers can be formed, some of which will be described below.
- Each of the layers may include one or more agents in the same or different proportions from layer to layer.
- the layers may be solid, porous, or filled with other drugs or excipients.
- FIG. 9 shows the simplest arrangement of layers including identical layers 50 that together form a uniform, homogeneous distribution of beneficial agent. If the carrier polymer were comprised of a biodegradable material, then erosion of the beneficial agent containing carrier would occur on both faces of the opening at the same time, and beneficial agent would be released at an approximately linear rate over time corresponding to the erosion rate of the carrier. This linear or constant release rate is referred to as a zero order delivery profile.
- Use of biodegradable carriers in combination with through openings is especially useful, to guarantee 100% discharge of the beneficial agent within a desired time without creating virtual spaces or voids between the radially outermost surface of the stent and tissue of the vessel wall.
- the openings may provide a communication between the strut-covered vessel wall and the blood stream. Such communication may accelerate vessel healing and allow the ingrowth of cells and extracellular components that more thoroughly lock the stent in contact with the vessel wall.
- some through-openings may be loaded with beneficial agent while others are left unloaded. The unloaded holes could provide an immediate nidus for the ingrowth of cells and extracellular components to lock the stent into place, while loaded openings dispense the beneficial agent.
- a stent based therapy using through openings, biodegradable carriers, and associated techniques described herein could be used to deliver a chemically ablative agent in a specific, precise pattern to a specific area for treatment of atrial fibrillation, while guaranteeing that none of the inherently cytotoxic ablating agent could be permanently trapped in contact with the tissue of the vessel wall.
- beneficial agents located in openings provide an equally dramatic advantage over surface coated devices.
- a composition comprising a beneficial agent and a non-biodegradable carrier would be loaded into the through openings, preferably in combination with a diffusion barrier layer as described below.
- a diffusion barrier layer as described below.
- C is the concentration of beneficial agent at cross section x
- x is either the thickness of a surface coating or depth of a through opening
- D is the diffusion coefficient
- t is time.
- the ratio of diffusion times to achieve comparable concentrations thus varies as the square of the ratio of depths.
- a typical opening depth is about 140 microns while a typical coating thickness is about 5 micron; the square of this ratio is 784, meaning that the effective duration of therapy for through openings is potentially almost three orders of magnitude greater for through openings than for surface coatings of the same composition.
- the inherent non-linearity of such release profiles can in part be compensated for in the case of through openings, but not in thin surface coatings, by varying the beneficial agent concentration of layers in a through opening as described below.
- Beneficial agent that is released to the radially innermost or inwardly facing surface known as the lumen facing surface of an expanded device may be rapidly carried away from the targeted area, for example by the bloodstream, and thus lost. Up to half of the total agent loaded in such situations may have no therapeutic effect due to being carried away by the bloodstream. This is probably the case for all surface coated stents as well as the through opening device of FIG. 9.
- FIG. 10 shows a device in which the first layer 52 is loaded into a through opening 24 such that the inner surface of the layer is substantially co-planar with the inwardly facing surface 54 of the cylindrical device.
- the first layer 52 is comprised of a material called a barrier material which blocks or retards biodegradation of subsequent layers in the inwardly facing direction toward the vessel lumen, and/or blocks or retards diffusion of the beneficial agent in that direction. Biodegradation of other layers or beneficial agent diffusion can then proceed only in the direction of the outwardly facing surface 56 of the device, which is in direct contact with the targeted tissue of the vessel wall.
- the barrier layer 52 may also function to prevent hydration of inner layers of beneficial agent and thus prevent swelling of the inner layers when such layers are formed of hygroscopic materials.
- the barrier layer 52 may further be comprised of a biodegradable material that degrades at a much slower rate than the biodegradable material in the other layers, so that the opening will eventually be entirely cleared. Providing a barrier layer 52 in the most inwardly facing surface of a through-opening thus guarantees that the entire load of beneficial agent is delivered to the target area in the vessel wall.
- Barrier layers can be used to control beneficial agent release kinetics in more sophisticated ways.
- a barrier layer 52 with a pre-determined degradation time could be used to deliberately terminate the beneficial agent therapy at a pre-determined time, by exposing the underlying layers to more rapid bio-degradation from both sides.
- Barrier layers can also be formulated to be activated by a separate, systemically applied agent. Such systemically applied agent could change the porosity of the barrier layer and/or change the rate of bio-degradation of the barrier layer or the bulk beneficial agent carrier.
- release of the beneficial agent could be activated by the physician at will by delivery of the systemically applied agent.
- a further embodiment of physician activated therapy would utilize a beneficial agent encapsulated in micro-bubbles and loaded into device openings.
- ultrasonic energy from an exterior of the body could be used to collapse the bubbles at a desired time, releasing the beneficial agent to diffuse to the outwardly facing surface of the reservoirs.
- These activation techniques can be used in conjunction with the release kinetics control techniques described herein to achieve a desired drug release profile that can be activated and/or terminated at selectable points in time.
- FIG. 11A shows an arrangement of layers provided in a through opening in which layers 50 of a beneficial agent in a biodegradable carrier material, are alternated with layers 58 of the biodegradable carrier material alone, with no active agent loaded, and a barrier layer 52 is provided at the inwardly facing surface.
- a beneficial agent in a biodegradable carrier material is alternated with layers 58 of the biodegradable carrier material alone, with no active agent loaded, and a barrier layer 52 is provided at the inwardly facing surface.
- FIG. 11B shows an arrangement of layers provided in a through opening in which layers 50 of a beneficial agent in a biodegradable carrier material, are alternated with layers 58 of the biodegradable carrier material alone, with no active agent loaded, and a barrier layer 52 is provided at the inwardly facing surface.
- FIG. 11B shows an arrangement of layers provided in a through opening in which layers 50 of a beneficial agent in a biodegradable carrier material, are alternated with layers 58 of the biodegradable carrier material alone
- different layers could be comprised of different beneficial agents altogether, creating the ability to release different beneficial agents at different points in time, as shown in FIG. 12.
- a layer 60 of anti-thrombotic agent could be deposited at the inwardly facing surface of the stent, followed by a barrier layer 52 and alternating layers of anti-proliferatives 62 and anti-inflamatories 64 .
- This configuration could provide an initial release of anti-thrombotic agent into the bloodstream while simultaneously providing a gradual release of anti-proliferatives interspersed with programmed bursts of anti-inflammatory agents to the vessel wall.
- the configurations of these layers can be designed to achieve the agent delivery bursts at particular points in time coordinated with the body's various natural healing processes.
- FIG. 13 A further alternative is illustrated in FIG. 13.
- the concentration of the same beneficial agent is varied from layer to layer, creating the ability to generate release profiles of arbitrary shape.
- Progressively increasing the concentration of agent in the layers 66 with increasing distance from the outwardly facing surface 56 for example, produces a release profile with a progressively increasing release rate, which would be impossible to produce in a thin surface coating.
- ⁇ N/ ⁇ t is the number of molecules per unit time
- A is the instantaneous drug eluting surface area
- D is the diffusivity
- C is the concentration.
- the drug eluting surface area of a surface coated stent is simply the surface area of the stent itself. Since this area is fixed, this method of controlling release kinetics is not available to surface coated devices. Through openings, however, present several possibilities for varying surface area as a function of time.
- beneficial agent is provided in the openings 24 in the form of microspheres, particles or the like. Individual layers 70 can then be created that contain these particles. Further, the particle size can be varied from layer to layer. For a given layer volume, smaller particle sizes increase the total particle surface area in that layer, which has the effect of varying the total surface area of the beneficial agent from layer to layer. Since the flux of drug molecules is proportional to surface area, the total drug flux can be adjusted from layer to layer by changing the particle size, and the net effect is control of release kinetics by varying particle sizes within layers.
- FIG. 15A shows an opening 70 having a conical shape cut into the material of the stent itself.
- the opening 70 may then be filled with beneficial agent 72 in layers as described above or in another manner.
- a barrier layer 74 may be provided on the inwardly facing side of the opening 70 to prevent the beneficial agent 72 from passing into the blood stream.
- the drug eluting surface area A t would continuously diminish (from FIG. 15A to FIG. 15B) as the bio-degradable carrier material erodes, yielding the elution pattern of FIG. 15C.
- FIG. 16A shows a simple cylindrical through-opening 80 in which a preformed, inverted cone 82 of beneficial agent has been inserted.
- the rest of the through opening 80 is then back-filled with a biodegradable substance 84 with a much slower rate of degradation or a non-biodegradable substance, and the inwardly facing opening of the through opening is sealed with a barrier layer 86 .
- This technique yields the opposite behavior to the previous example.
- the drug-eluting surface area A t continuously increases with time between FIGS. 16A and 16B, yielding the elution pattern of FIG. 16C.
- the changing cross section openings 70 of FIG. 15A and the non-biodegradable backfilling techniques of FIG. 16A may be combined with any of the layered agent embodiments of FIGS. 9-14 to achieve desired release profiles.
- the embodiment of FIG. 15A may use the varying agent concentration layers of FIG. 13 to more accurately tailor a release curve to a desired profile.
- FIG. 17A shows a through opening 90 in which a spherical beneficial agent plug 92 has been inserted.
- the resulting biodegradation of the sphere in which the cross sectional surface area varies as a sinusoidal function of depth, produces a flux density which is roughly a sinusoidal function of time, FIG. 17B.
- Other results are of course possible with other profiles, but none of these more complex behaviors could be generated in a thin, fixed-area surface coating.
- FIGS. 18A-18C use a barrier layer 52 ′ with an opening 96 to achieve the increasing agent release profile of FIG. 16C.
- the opening 24 is provided with an inner barrier layer 52 and multiple beneficial agent layers 50 as in the embodiment of FIG. 10.
- An additional outer barrier layer 52 ′ is provided with a small hole 96 for delivery of the agent to the vessel wall.
- the beneficial agent containing layers 50 degrade in a hemispherical pattern resulting in increasing surface area for agent delivery over time and thus, an increasing agent release profile.
- FIG. 19 illustrates an alternative embodiment in which an opening in the tissue supporting device is loaded with cylindrical layers of beneficial agent.
- the entire device is coated with sequential layers 100 , 102 , 104 , 106 of beneficial agent.
- the interior surface 54 and exterior surface 56 of the device are then stripped to remove the beneficial agent on these surfaces leaving the cylindrical layers of beneficial agent in the openings.
- a central opening remains after the coating layers have been deposited which allows communication between the outer surface 56 and inner surface 54 of the tissue supporting device.
- the cylindrical layers are eroded sequentially. This can be used for pulsatile delivery of different beneficial agents, delivery of different concentrations of beneficial agents, or delivery of the same agent.
- the ends of the cylindrical layers 100 , 102 , 104 , 106 are exposed. This results in a low level of erosion of the underlying layers during erosion of an exposed layer.
- the ends of the cylindrical layers may be covered by a barrier layer to prevent this low level continuous erosion. Erosion rates of the cylindrical layers may be further controlled by contouring the surfaces of the layers.
- a ribbed or star-shaped pattern may be provided on the radially inner layers to provide a uniform surface area or uniform erosion rate between the radially inner layers and the radially outer layers. Contouring of the surfaces of layers may also be used in other embodiments to provide an additional variable for controlling the erosion rates.
- FIG. 20 illustrates a further alternative embodiment of the invention in which different beneficial agents are positioned in different holes of an expandable medical device 300 .
- a first beneficial agent is provided in holes 330 a at the ends of the device and a second beneficial agent is provided in holes 330 b at a central portion of the device.
- the beneficial agent may contain different drugs, the same drugs in different concentrations, or different variations of the same drug.
- the embodiment of FIG. 20 may be used to provide an expandable medical device 300 with either “hot ends” or “cool ends.”
- each end portion of the device 300 which includes the holes 330 a containing the first beneficial agent extends at least one hole and up to about 15 holes from the edge. This distance corresponds to about 0.005 to about 0.1 inches from the edge of an unexpanded device.
- the distance from the edge of the device 300 which includes the first beneficial agent is preferably about one section, where a section is defined between the bridging elements.
- Different beneficial agents containing different drugs may be disposed in different openings in the stent. This allows the delivery of two or more beneficial agents from a single stent in any desired delivery pattern. Alternatively, different beneficial agents containing the same drug in different concentrations may be disposed in different openings. This allows the drug to be uniformly distributed to the tissue with a non-uniform device structure.
- the two or more different beneficial agents provided in the devices described herein may contain (1) different drugs; (2) different concentrations of the same drug; (3) the same drug with different release kinetics, i.e., different matrix erosion rates; or (4) different forms of the same drug.
- Examples of different beneficial agents formulated containing the same drug with different release kinetics may use different carriers to achieve the elution profiles of different shapes.
- Some examples of different forms of the same drug include forms of a drug having varying hydrophilicity or lipophilicity.
- the holes 330 a at the ends of the device are loaded with a first beneficial agent comprising a drug with a high lipophilicity while holes 330 b at a central portion of the device are loaded with a second beneficial agent comprising the drug with a lower lipophilicity.
- the first high lipophilicity beneficial agent at the “hot ends” will diffuse more readily into the surrounding tissue reducing the edge effect restenosis.
- the device 300 may have an abrupt transition line at which the beneficial agent changes from a first agent to a second agent. For example, all openings within 0.05 inches of the end of the device may contain the first agent while the remaining openings contain the second agent. Alternatively, the device may have a gradual transition between the first agent and the second agent. For example, a concentration of the drug in the openings can progressively increase (or decrease) toward the ends of the device. In another example, an amount of a first drug in the openings increases while an amount of a second drug in the openings decreases moving toward the ends of the device.
- FIG. 21 illustrates a further alternative embodiment of an expandable medical device 400 in which different beneficial agents are positioned in different openings 430 a , 430 b in the device in an alternating or interspersed manner.
- multiple beneficial agents can be delivered to tissue over the entire area or a portion of the area supported by the device.
- This embodiment will be useful for delivery of multiple beneficial agents where combination of the multiple agents into a single composition for loading in the device is not possible due to interactions or stability problems between the beneficial agents.
- the loading of different beneficial agents in different openings may be used to provide a more even spatial distribution of the beneficial agent delivered in instances where the expandable medical device has a non-uniform distribution of openings in the expanded configuration.
- the coverage of the device 500 is greater at the cylindrical tube portions 512 of the device than at the bridging elements 514 .
- Coverage is defined as the ratio of the device surface area to the area of the lumen in which the device is deployed.
- the concentration of the beneficial agent may be varied in the openings at portions of the device to achieve a more even distribution of the beneficial agent throughout the tissue.
- the openings 530 a in the tube portions 512 include a beneficial agent with a lower drug concentration than the openings 530 b in the bridging elements 514 .
- the uniformity of agent delivery may be achieved in a variety of manners including varying the drug concentration, the opening diameter or shape, the amount of agent in the opening (i.e., the percentage of the opening filed), the matrix material, or the form of the drug.
- beneficial agents may be delivered to the lumenal side of the expandable medical device.
- Drugs which are delivered into the blood stream from the lumenal side of the device can be located at a proximal end of the device or a distal end of the device.
- the methods for loading different beneficial agents into different openings in an expandable medical device may include known techniques such as dipping and coating and also known piezoelectric micro-jetting techniques.
- Micro-injection devices may be computer controlled to deliver precise amounts of two or more liquid beneficial agents to precise locations on the expandable medical device in a known manner.
- a dual agent jetting device may deliver two agents simultaneously or sequentially into the openings.
- a lumenal side of the through openings may be blocked during loading by a resilient mandrel allowing the beneficial agents to be delivered in liquid form, such as with a solvent.
- the beneficial agents may also be loaded by manual injection devices.
- the therapeutic agent layers of the present invention are beneficial agents comprised of a matrix and at least one therapeutic agent.
- matrix or “biocompatible matrix” are used interchangeably to refer to a medium or material that, upon implantation in a subject, does not elicit a detrimental response sufficient to result in the rejection of the matrix.
- the matrix typically does not provide any therapeutic responses itself, though the matrix may contain or surround a therapeutic agent, a therapeutic agent, an activating agent or a deactivating agent, as defined herein.
- a matrix is also a medium that may simply provide support, structural integrity or structural barriers.
- the matrix may be polymeric, non-polymeric, hydrophobic, hydrophilic, lipophilic, amphiphilic, and the like.
- the matrix of the therapeutic agent layers can be made from pharmaceutically acceptable polymers, such as those typically used in medical devices.
- polymer refers to molecules formed from the chemical union of two or more repeating units, called monomers. Accordingly, included within the term “polymer” may be, for example, dimers, trimers and oligomers.
- the polymer may be synthetic, naturally-occurring or semisynthetic.
- the term “polymer” refers to molecules which typically have a M w greater than about 3000 and preferably greater than about 10,000 and a M w that is less than about 10 million, preferably less than about a million and more preferably less than about 200,000.
- polymers include but are not limited to, poly- ⁇ -hydroxy acid esters such as, polylactic acid, polyglycolic acid, polylactic-co-glycolic acid, polylactic acid-co-caprolactone; polyethylene glycol and polyethylene oxide; polyvinyl pyrrolidone; polyorthoesters; polysaccharides and polysaccharide derivatives such as polyhyaluronic acid, polyalginic acid, chitin, chitosan, cellulose, hydroxyehtylcellulose, hydroxypropylcellulose, carboxymethylcellulose; polypeptides, and proteins such as polylysine, polyglutamic acid, albumin; polyanhydrides; polyhydroxy alkonoates such as polyhydroxy valerate, polyhydroxy butyrate, and the like, and copolymers thereof.
- poly- ⁇ -hydroxy acid esters such as, polylactic acid, polyglycolic acid, polylactic-co-glycolic acid, polylactic acid-co-caprolactone
- the polymers and copolymers can be prepared by methods well known in the art (see, for example, Rempp and Merril: Polymer Synthesis, 1998, John Wiley and Sons) in or can be used as purchased from Alkermes, in Cambridge, Mass. or Birmingham Polymer Inc., in Birmingham, Ala.
- the preferred co-polymer for use in the present invention are poly(lactide-co-glycolide) (PLGA) polymers.
- the rate at which the polymer erodes is determined by the selection of the ratio of lactide to glycolide within the copolymer, the molecular weight of each polymer used, and the crystallinity of the polymers used.
- Bioerodible polymers may also be used to form barrier layers that erode at a rate that can be predetermined base on the composition and that contain no therapeutic agent.
- Typical additives that may be included in a bioerodible matrix are well known to those skilled in the art (see Remington: The Science and Practice of Pharmacy, Gennaro, ed., Mack Publishing Co., Easton, Pa., 19th ed., 1995) and include but are not limited to pharmaceutically acceptable excipients, adjuvants, carriers, antioxidants, preservatives, buffers, antacids, emulsifiers, inert fillers, fragrances, thickeners, tackifiers, opacifiers, gelling agents, stabilizers, surfactants, emolliehts, coloring agents, and the like.
- Typical formulations for therapeutic agents incorporated in these medical devices are well known to those skilled in the art and include but are not limited to solid particle dispersions, encapsulated agent dispersions, and emulsions, suspensions, liposomes or microparticles, wherein said liposome or microparticle comprise a homogeneous or heterogeneous mixture of the therapeutic agent.
- homogeneously disposed refers to a component which is mixed uniformly in a matrix in such a manner that the component is macroscopically indistinguishable from the matrix itself.
- a homogeneously disposed component is a drug formulation such as a microemulsion in which small beads of oil are dispersed uniformly in water.
- heterogeneously disposed refers to a component which is mixed non-uniformly into a matrix in such a manner that the component is macroscopically distinguishable from the matrix itself.
- An example of a heterogeneously disposed component is a simple emulsion in which the beads of oil in the water are large enough to cause a turbidity to the solution and can be seen settling out of solution over time.
- Heterogeneously disposed compositions also include encapsulated formulations where a component, such as a protective layer, is layered onto or around a therapeutic agent or a therapeutic layer, forming a protective shell.
- the amount of the drug that is present in the device, and that is required to achieve a therapeutic effect depends on many factors, such as the minimum necessary dosage of the particular drug, the condition to be treated, the chosen location of the inserted device, the actual compound administered, the age, weight, and response of the individual patient, the severity of the patient's symptoms, and the like.
- the appropriate dosage level of the therapeutic agent for more traditional routes of administration, are known to one skilled in the art. These conventional dosage levels correspond to the upper range of dosage levels for compositions, including a physiologically active substance and traditional penetration enhancer. However, because the delivery of the active substance occurs at the site where the drug is required, dosage levels significantly lower than a conventional dosage level may be used with success.
- the percentage of therapeutic agent in the composition is determined by the required effective dosage, the therapeutic activity of the particular formulation, and the desired release profile.
- the active substance will be present in the composition in an amount from about 0.0001% to about 99%, more preferably about 0.01% to about 80% by weight of the total composition depending upon the particular substance employed. However, generally the amount will range from about 0.01% to about 75% by weight of the total composition, with levels of from about 25% to about 75% being preferred.
- the protective and barrier layers of the present invention are beneficial agents comprised of a bioerodible matrix and optionally contain additional additives, therapeutic agents, activating agents, deactivating agents, and the like. Either a property of the chosen material of the protective or barrier layer, or a chemical embedded in the protective or barrier layer provides protection from deactivating processes or conditions for at least one therapeutic agent.
- the protective or barrier layer may also be comprised of pharmaceutically acceptable lipids or lipid derivatives, which are well known in the art and include but are not limited to fatty acids, fatty acid esters, lysolipids, phosphocholines, (Avanti Polar Lipids, Alabaster, Ala.), including 1-alkyl-2-acetoyl-sn-glycero 3-phosphocholines, and 1-alkyl-2-hydroxy-sn-glycero 3-phosphocholines; phosphatidylcholine with both saturated and unsaturated lipids, including dioleoylphosphatidylcholine; dimyristoyl-phosphatidylcholine; dipentadecanoylphosphatidylcholine; dilauroylphosphatidyl-choline; dipalmitoylphosphatidylcholine (DPPC); distearoylphosphatidylcholine (DSPC); and diarachidonylphosphatidyl
- a cationic lipid may be used, such as, for example, N-[1-(2,3-dioleoyloxy)propyl]-N,N,N-trimethylammonium chloride (DOTMA), 1,2-dioleoyloxy-3-(trimethylammonio)propane (DOTAP); and 1,2-dioleoyl-3-(4′-trimethylammonio)butanoyl-sn-glycerol (DOTB).
- DOTMA 1,2-dioleoyloxy-3-(trimethylammonio)propane
- DOB 1,2-dioleoyl-3-(4′-trimethylammonio)butanoyl-sn-glycerol
- the molar ratio of cationic lipid to non-cationic lipid may be, for example, from about 1:1000 to about 1:100.
- the molar ratio of cationic lipid to non-cationic lipid may be from about 1:2 to about 1:10, with a ratio of from about 1:1 to about 1:2.5 being preferred. Even more preferably, the molar ratio of cationic lipid to non-cationic lipid may be about 1:1.
- the preferred lipids for use in the present invention are phosphatidyl-choline, phosphatidylethanolamine, phosphatidylserine, sphingomyelin as well as synthetic phospholipids such as dimyristoyl phosphatidylcholine, dipalmitoyl phosphatidylcholine, distearoyl phosphatidylcholine, distearoyl phosphatidyl-glycerol, dipalmitoyl phosphatidylglycerol, dimyristoyl phosphatidylserine, distearoyl phosphatidylserine and dipalmitoyl phosphatidylserine.
- the rate at which the bioerodible matrix erodes is determined by the choice of lipid, the molecular weight, and the ratio of the chosen materials.
- the protective or barrier layer can erode by either chemical or physical erosion mechanisms. If the layer erodes by a physical mechanism, the layer is typically a thin film from about 0.1 ⁇ m to about 3 ⁇ m of a non-polymeric material embedded between two polymeric layers. In this instance, the structural integrity of the protective or barrier layer is maintained by the presence of both of these polymeric layers. When the polymeric material closest to the luminal surface erodes away, the protective or barrier layer breaks apart by the physical forces exerted on it from the remaining polymeric layer. In another embodiment, the protective or barrier layer is eroded by chemical interactions, dissolution in water, hydrolysis, or reaction with enzymes.
- One function of the protective or barrier layer is to protect one or more therapeutic agents from deactivating or degrading conditions.
- the protection may come from the properties of the material when, for example, a hydrophobic protective or barrier layer would protect a water sensitive agent from water by resisting the influx of moisture.
- the protective or barrier layer may also act as a physical barrier.
- a protective or barrier layer comprised of a hydrogel may allow water to be absorbed by the gel, and allow any agents contained within the gel to diffuse out of the gel into the reaction environment. The hydrogel, however, would prevent enzymes from penetrating the layer, thereby protecting any agents contained within from the enzyme.
- the term “hydrogel” refers to cross-linked polymeric material in which the liquid component is water. Hydrogels may be prepared by cross-linking certain polymers and lipids disclosed herein.
- the protective or barrier layer does not have to act as a barrier.
- the protective or barrier layer may protect a therapeutic agent by releasing an agent, such as an activating agent or a deactivating agent, into the reaction environment prior to the release of the therapeutic agent.
- a therapeutic agent may be incorporated directly in the protective or barrier layer.
- the therapeutic agent can be heterogeneously or homogeneously dispersed in the protective or barrier layer.
- the therapeutic agent can be a drug, or a drug formulated into a microcapsule, niosome, liposome, microbubble, microsphere, or the like.
- the protective or barrier layer may contain more than one therapeutic agent.
- a water sensitive drugs such as a limus, or any other drug that must be administered through intravenous, intramuscular, or subcutaneously, could be incorporated in a hydrophobic matrix such as SAIB, or fatty acid ester.
- a therapeutic agent may also be disposed in a therapeutic agent layer, separate from the protective or barrier layer.
- the protective or barrier layer may be adjacent to the therapeutic agent layer and may serve to prevent or retard processes that would degrade or deactivate the therapeutic agent until the protective or barrier layer has substantially eroded.
- the protective or barrier layer is a barrier between a therapeutic layer and the reaction environment. This barrier may be a hydrophobic barrier that resists water absorption. The hydrophobic barrier would be used in conjunction with water-sensitive drugs as described above.
- the protective or barrier layer maybe a hydrogel that resists the absorbance of enzymes. An enzyme resistant barrier would used to protect an drug such as a DNA, RNA, peptide or protein based therapeutic agent.
- the protective or barrier layer may optionally include activating and deactivating agents for the purpose of preparing the reaction environment for the subsequent release of a therapeutic agent.
- activating and deactivating agents are well known to those skilled in the art and include but are not limited to antacids, buffers, enzyme inhibitors, hydrophobic additives, and adjuvants.
- Mg(OH) 2 in particles of about 0.5 ⁇ m to about 5 ⁇ m more preferably, about 1 ⁇ m incorporated in a PLGA polymer layer could be used in conjunction with any acid senstive drug.
- An example of an activating agent is chymotrypsin, which may be incorporated in polyvinyl pyrrolidone layer. The chymotrypsin, could be used to convert a pro-drug to an active drug.
Abstract
Description
- This application is a continuation-in-part of pending U.S. application Ser. No. 10/668,430, filed Sep. 22, 2003, which claims priority to U.S. Provisional Application Ser. No. 60/412,489, filed Sep. 20, 2002. This application is also a continuation-in-part of pending U.S. application Ser. No. 10/253,020, filed on Sep. 23, 2002, which is a continuation-in-part of U.S. application Ser. No. 09/948,989, filed on Sep. 7, 2001, which claims priority to U.S. Provisional Application Ser. No. 60/314,259, filed Aug. 20, 2001 and which is a continuation-in-part of U.S. application Ser. No. 09/688,092, filed Oct. 16, 2000 which is a continuation-in-part of U.S. application Ser. No. 09/183,555, filed Oct. 29, 1998, now U.S. Pat. No. 6,241,762, which claims priority to U.S. Provisional Application Ser. No. 60/079,881, filed Mar. 30, 1998.
- 1. Field of the Invention
- The present invention relates to tissue-supporting medical devices, and more particularly to expandable, non-removable devices that are implanted within a bodily lumen of a living animal or human to support the organ and maintain patency, and that can deliver a beneficial agent to the intervention site.
- 2. Summary of the Related Art
- In the past, permanent or biodegradable devices have been developed for implantation within a body passageway to maintain patency of the passageway. These devices are typically introduced percutaneously, and transported transluminally until positioned at a desired location. These devices are then expanded either mechanically, such as by the expansion of a mandrel or balloon positioned inside the device, or expand themselves by releasing stored energy upon actuation within the body. Once expanded within the lumen, these devices, called stents, become encapsulated within the body tissue and remain a permanent implant.
- Known stent designs include monofilament wire coil stents (U.S. Pat. No. 4,969,458); welded metal cages (U.S. Pat. Nos. 4,733,665 and 4,776,337); and, most prominently, thin-walled metal cylinders with axial slots formed around the circumference (U.S. Pat. Nos. 4,733,665; 4,739,762; and 4,776,337). Known construction materials for use in stents include polymers, organic fabrics and biocompatible metals, such as, stainless steel, gold, silver, tantalum, titanium, and shape memory alloys such as Nitinol.
- U.S. Pat. Nos. 4,733,665; 4,739,762; and 4,776,337 disclose expandable and deformable interluminal vascular grafts in the form of thin-walled tubular members with axial slots allowing the members to be expanded radially outwardly into contact with a body passageway. After insertion, the tubular members are mechanically expanded beyond their elastic limit and thus permanently fixed within the body. U.S. Pat. No. 5,545,210 discloses a thin-walled tubular stent geometrically similar to those discussed above, but constructed of a nickel-titanium shape memory alloy (“Nitinol”), which can be permanently fixed within the body without exceeding its elastic limit. All of these stents share a critical design property: in each design, the features that undergo permanent deformation during stent expansion are prismatic, i.e., the cross sections of these features remain constant or change very gradually along their entire active length. These prismatic structures are ideally suited to providing large amounts of elastic deformation before permanent deformation commences, which in turn leads to sub-optimal device performance in important properties including stent expansion force, stent recoil, strut element stability, stent securement on delivery catheters, and radiopacity.
- U.S. Pat. No. 6,241,762, which is incorporated herein by reference in its entirety, discloses a non-prismatic stent design which remedies the above mentioned performance deficiencies of previous stents. In addition, preferred embodiments of this patent provide a stent with large, non-deforming strut and link elements, which can contain holes without compromising the mechanical properties of the strut or link elements, or the device as a whole. Further, these holes may serve as large, protected reservoirs for delivering various beneficial agents to the device implantation site.
- Of the many problems that may be addressed through stent-based local delivery of beneficial agents, one of the most important is restenosis. Restenosis is a major complication that can arise following vascular interventions such as angioplasty and the implantation of stents. Simply defined, restenosis is a wound healing process that reduces the vessel lumen diameter by extracellular matrix deposition and vascular smooth muscle cell proliferation, and which may ultimately result in renarrowing or even reocclusion of the lumen. Despite the introduction of improved surgical techniques, devices and pharmaceutical agents, the overall restenosis rate is still reported in the range of 25% to 50% within six to twelve months after an angioplasty procedure. To treat this condition, additional revascularization procedures are frequently required, thereby increasing trauma and risk to the patient.
- Some of the techniques under development to address the problem of restenosis include irradiation of the injury site and the use of conventional stents to deliver a variety of beneficial or pharmaceutical agents to the wall of the traumatized vessel. In the latter case, a conventional stent is frequently surface-coated with a beneficial agent (often a drug-impregnated polymer) and implanted at the angioplasty site. Alternatively, an external drug-impregnated polymer sheath is mounted over the stent and co-deployed in the vessel.
- While acute outcomes from radiation therapies appeared promising initially, long term beneficial outcomes have been limited to reduction in restenosis occurring within a previously implanted stent, so-called ‘in-stent’ restenosis. Radiation therapies have not been effective for preventing restenosis in de novo lesions. Polymer sheaths that span stent struts have also proven problematic in human clinical trials due to the danger of blocking flow to branch arteries, incomplete apposition of stent struts to arterial walls and other problems. Unacceptably high levels of MACE (Major Adverse Cardiac Events that include death, heart attack, or the need for a repeat angioplasty or coronary artery bypass surgery) have resulted in early termination of clinical trials for sheath covered stents.
- Conventional stents with surface coatings of various beneficial agents, by contrast, have shown promising early results. U.S. Pat. No. 5,716,981, for example, discloses a stent that is surface-coated with a composition comprising a polymer carrier and paclitaxel (a well-known compound that is commonly used in the treatment of cancerous tumors). The patent offers detailed descriptions of methods for coating stent surfaces, such as spraying and dipping, as well as the desired character of the coating itself: it should “coat the stent smoothly and evenly” and “provide a uniform, predictable, prolonged release of the anti-angiogenic factor.” Surface coatings, however, can provide little actual control over the release kinetics of beneficial agents. These coatings are necessarily very thin, typically 5 to 8 microns deep. The surface area of the stent, by comparison is very large, so that the entire volume of the beneficial agent has a very short diffusion path to discharge into the surrounding tissue.
- Increasing the thickness of the surface coating has the beneficial effects of improving drug release kinetics including the ability to control drug release and to allow increased drug loading. However, the increased coating thickness results in increased overall thickness of the stent wall. This is undesirable for a number of reasons, including increased trauma to the vessel wall during implantation, reduced flow cross-section of the lumen after implantation, and increased vulnerability of the coating to mechanical failure or damage during expansion and implantation. Coating thickness is one of several factors that affect the release kinetics of the beneficial agent, and limitations on thickness thereby limit the range of release rates, durations, and the like that can be achieved.
- In addition to sub-optimal release profiles, there are further problems with surface coated stents. The fixed matrix polymer carriers frequently used in the device coatings typically retain approximately 30% of the beneficial agent in the coating indefinitely. Since these beneficial agents are frequently highly cytotoxic, sub-acute and chronic problems such as chronic inflammation, late thrombosis, and late or incomplete healing of the vessel wall may occur. Additionally, the carrier polymers themselves are often highly inflammatory to the tissue of the vessel wall. On the other hand, use of bio-degradable polymer carriers on stent surfaces can result in the creation of “virtual spaces” or voids between the stent and tissue of the vessel wall after the polymer carrier has degraded, which permits differential motion between the stent and adjacent tissue. Resulting problems include micro-abrasion and inflammation, stent drift, and failure to re-endothelialize the vessel wall.
- Another significant problem is that expansion of the stent may stress the overlying polymeric coating causing the coating to plastically deform or even to rupture, which may therefore effect drug release kinetics or have other untoward effects. Further, expansion of such a coated stent in an atherosclerotic blood vessel will place circumferential shear forces on the polymeric coating, which may cause the coating to separate from the underlying stent surface. Such separation may again have untoward effects including embolization of coating fragments causing vascular obstruction.
- In view of the drawbacks of the prior art, it would be advantageous to provide a stent capable of delivering a relatively large volume of a beneficial agent to a traumatized site in a vessel while avoiding the numerous problems associated with surface coatings containing beneficial agents, without increasing the effective wall thickness of the stent, and without adversely impacting the mechanical expansion properties of the stent.
- It would further be advantageous to have such a stent, which also significantly increases the available depth of the beneficial agent reservoir.
- It would also be advantageous to have methods of loading various beneficial agents or combinations of beneficial agents into these deep reservoirs, which provided control over the temporal release kinetics of the agents.
- In accordance with one aspect of the invention, a device for the controlled release of one or more drugs comprises an implantable stent, at least two reservoirs in the stent, and a release system contained in each of the at least two reservoirs, wherein the release system comprises one or more drugs for release.
- The invention will now be described in greater detail with reference to the preferred embodiments illustrated in the accompanying drawings, in which like elements bear like reference numerals, and wherein:
- FIG. 1 is a perspective view of a tissue supporting device in accordance with a first preferred embodiment of the present invention;
- FIG. 2 is an enlarged side view of a portion of the device of FIG. 1;
- FIG. 3 is an enlarged side view of a tissue supporting device in accordance with a further preferred embodiment of the present invention;
- FIG. 4 is an enlarged side view of a portion of the stent shown in FIG. 3;
- FIG. 5 is an enlarged cross section of an opening;
- FIG. 6 is an enlarged cross section of an opening illustrating beneficial agent loaded into the opening;
- FIG. 7 is an enlarged cross section of an opening illustrating a beneficial agent loaded into the opening and a thin coating of a beneficial agent;
- FIG. 8 is an enlarged cross section of an opening illustrating a beneficial agent loaded into the opening and thin coatings of different beneficial agents on different surfaces of the device;
- FIG. 9 is an enlarged cross section of an opening illustrating a beneficial agent provided in a plurality of layers;
- FIG. 10 is an enlarged cross section of an opening illustrating a beneficial agent and a barrier layer loaded into the opening in layers;
- FIG. 11A is an enlarged cross section of an opening illustrating a beneficial agent, a biodegradable carrier, and a barrier layer loaded into the opening in layers;
- FIG. 11B is a graph of the release kinetics of the device of FIG. 11A;
- FIG. 12 is an enlarged cross section of an opening illustrating different beneficial agents, carrier, and barrier layers loaded into the opening;
- FIG. 13 is an enlarged cross section of an opening illustrating a beneficial agent loaded into the opening in layers of different concentrations;
- FIG. 14 is an enlarged cross section of an opening illustrating a beneficial agent loaded into the opening in layers of microspheres of different sizes;
- FIG. 15A is an enlarged cross section of a tapered opening illustrating a beneficial agent loaded into the opening;
- FIG. 15B is an enlarged cross section of the tapered opening of FIG. 15A with the beneficial agent partially degraded;
- FIG. 15C is a graph of the release kinetics of the device of FIGS. 15A and 15B;
- FIG. 16A is an enlarged cross section of an opening illustrating a beneficial agent loaded into the opening in a shape configured to achieve a desired agent delivery profile;
- FIG. 16B is an enlarged cross section of the opening of FIG. 16A with the beneficial agent partially degraded;
- FIG. 16C is a graph of the release kinetics of the device of FIGS. 16A and 16B;
- FIG. 17A is an enlarged cross section of an opening illustrating the beneficial agent loaded into the opening and a spherical shape;
- FIG. 17B is a graph of the release kinetics of the device of FIG. 17A;
- FIG. 18A is an enlarged cross section of an opening illustrating a plurality of beneficial agent layers and a barrier layer with an opening for achieving a desired agent delivery profile;
- FIG. 18B is an enlarged cross section of the opening of FIG. 18A with the agent layers beginning to degraded;
- FIG. 18C is an enlarged cross section of the opening of FIG. 18A with the agent layers further degraded;
- FIG. 19 is an enlarged cross section of an opening illustrating a plurality of cylindrical beneficial agent layers;
- FIG. 20 is an isometric view of an expandable tissue supporting device with different beneficial agents in different holes;
- FIG. 21 is an isometric view of an expandable tissue supporting device with different beneficial agents in alternating holes; and
- FIG. 22 is an enlarged side view of a portion of an expandable tissue supporting device with beneficial agent openings in the bridging elements.
- Referring to FIGS. 1 and 2, a tissue supporting device in accordance with one preferred embodiment of the present invention is shown generally by
reference numeral 10. Thetissue supporting device 10 includes a plurality ofcylindrical tubes 12 connected by S-shapedbridging elements 14. The bridgingelements 14 allow the tissue supporting device to bend axially when passing through the tortuous path of the vasculature to the deployment site and allow the device to bend when necessary to match the curvature of a vessel wall to be supported. Each of thecylindrical tubes 12 has a plurality ofaxial slots 16 extending from an end surface of the cylindrical tube toward an opposite end surface. - Formed between the
slots 16 is a network ofaxial struts 18 and links 22. Thestruts 18 andlinks 22 are provided with openings for receiving and delivering a beneficial agent. As will be described below with respect to FIGS. 9-17, the beneficial agent is loaded into the openings in layers or other configurations which provide control over the temporal release kinetics of the agent. - Each
individual strut 18 is preferably linked to the rest of the structure through a pair of reducedsections 20, one at each end, which act as stress/strain concentration features. The reducedsections 20 of the struts function as hinges in the cylindrical structure. Since the stress/strain concentration features are designed to operate into the plastic deformation range of generally ductile materials, they are referred to as ductile hinges 20. The ductile hinges 20 are described in further detail in U.S. Pat. No. 6,241,762, which has been incorporated herein by reference. - With reference to the drawings and the discussion, the width of any feature is defined as its dimension in the circumferential direction of the cylinder. The length of any feature is defined as its dimension in the axial direction of the cylinder. The thickness of any feature is defined as the wall thickness of the cylinder.
- The presence of the ductile hinges 20 allows all of the remaining features in the tissue supporting device to be increased in width or the circumferentially oriented component of their respective rectangular moments of inertia—thus greatly increasing the strength and rigidity of these features. The net result is that elastic, and then plastic deformation commence and propagate in the ductile hinges 20 before other structural elements of the device undergo any significant elastic deformation. The force required to expand the
tissue supporting device 10 becomes a function of the geometry of the ductile hinges 20, rather than the device structure as a whole, and arbitrarily small expansion forces can be specified by changing hinge geometry for virtually any material wall thickness. The ability to increase the width and thickness of thestruts 18 andlinks 22 provides additional area and depth for the beneficial agent receiving openings. - In the embodiment of FIGS. 1 and 2, it is desirable to increase the width of the individual struts 18 between the ductile hinges 20 to the maximum width that is geometrically possible for a given diameter and a given number of struts arrayed around that diameter. The only geometric limitation on strut width is the minimum practical width of the
slots 16 which is about 0.002 inches (0.0508 mm) for laser machining. Lateral stiffness of thestruts 18 increases as the cube of strut width, so that relatively small increases in strut width significantly increase strut stiffness. The net result of inserting ductile hinges 20 and increasing strut width is that thestruts 18 no longer act as flexible leaf springs, but act as essentially rigid beams between the ductile hinges. All radial expansion or compression of the cylindricaltissue supporting device 10 is accommodated by mechanical strain in the hinge features 20, and yield in the hinge commences at very small overall radial expansion or compression. - The
ductile hinge 20 illustrated in FIGS. 1 and 2 is exemplary of a preferred structure that will function as a stress/strain concentrator. Many other stress/strain concentrator configurations may also be used as the ductile hinges in the present invention, as shown and described by way of example in U.S. Pat. No. 6,241,762. The geometric details of the stress/strain concentration features or ductile hinges 20 can be varied greatly to tailor the exact mechanical expansion properties to those required in a specific application. - Although a tissue supporting device configuration has been illustrated in FIG. 1 which includes ductile hinges, it should be understood that the beneficial agent may be contained in openings in stents having a variety of designs including the designs illustrated in U.S. Provisional Patent Application Ser. No. 60/314,360, filed on Aug. 20, 2001 and U.S. patent application Ser. No. 09/948,987, filed on Sep. 7, 2001 (Attorney Docket No. 032304-033), which are incorporated herein by reference. The present invention incorporating beneficial agent openings may also be used with other known stent designs.
- As shown in FIGS. 1-4, at least one and more preferably a series of
openings 24 are formed by laser drilling or any other means known to one skilled in the art at intervals along the neutral axis of thestruts 18. Similarly, at least one and preferably a series ofopenings 26 are formed at selected locations in thelinks 22. Although the use ofopenings struts 18 andlinks 22 is preferred, it should be clear to one skilled in the art that openings could be formed in only one of the struts and links. Openings may also be formed in thebridging elements 14. In the embodiment of FIGS. 1 and 2, theopenings tissue supporting device 10. It should be apparent to one skilled in the art, however, that openings of any geometrical shape or configuration could of course be used without departing from the scope of the present invention. In addition, openings having a depth less than the thickness of the device may also be used. - The behavior of the
struts 18 in bending is analogous to the behavior of an I-beam or truss. Theouter edge elements 32 of thestruts 18, shown in FIG. 2, correspond to the I-beam flange and carry the tensile and compressive stresses, whereas theinner elements 34 of thestruts 18 correspond to the web of an I-beam which carries the shear and helps to prevent buckling and wrinkling of the faces. Since most of the bending load is carried by theouter edge elements 32 of thestruts 18, a concentration of as much material as possible away from the neutral axis results in the most efficient sections for resisting strut flexure. As a result, material can be judiciously removed along the axis of the strut so as to formopenings struts 18 andlinks 22 thus formed remain essentially rigid during stent expansion, theopenings - The
openings struts 18 may promote the healing of the intervention site by promoting regrowth of the endothelial cells. By providing theopenings openings - The
openings tissue supporting device 10 is supporting. - The terms “agent” and “beneficial agent” as used herein are intended to have their broadest possible interpretation and are used to include any therapeutic agent or drug, as well as inactive agents such as barrier layers or carrier layers. The terms “drug” and “therapeutic agent” are used interchangeably to refer to any therapeutically active substance that is delivered to a bodily conduit of a living being to produce a desired, usually beneficial, effect. The present invention is particularly well suited for the delivery of antiproliferatives (anti-restenosis agents) such as paclitaxel and rapamycin for example, and antithrombins such as heparin, for example.
- Additional uses, however, include therapeutic agents in all the major therapeutic areas including, but not limited to: anti-infectives such as antibiotics and antiviral agents; analgesics, including fentanyl, sufentanil, buprenorphine and analgesic combinations; anesthetics; anorexics; antiarthritics; antiasthmatic agents such as terbutaline; anticonvulsants; antidepressants; antidiabetic agents; antidiarrheals; antihistamines; anti-inflammatory agents; antimigraine preparations; antimotion sickness preparations such as scopolamine and ondansetron; antinauseants; antineoplastics; antiparkinsonism drugs; antipruritics; antipsychotics; antipyretics; antispasmodics, including gastrointestinal and urinary; anticholinergics; sympathomimetrics; xanthine derivatives; cardiovascular preparations, including calcium channel blockers such as nifedipine; beta blockers; beta-agonists such as dobutamine and ritodrine; antiarrythmics; antihypertensives such as atenolol; ACE inhibitors such as ranitidine; diuretics; vasodilators, including general, coronary, peripheral, and cerebral; central nervous system stimulants; cough and cold preparations; decongestants; diagnostics; hormones such as parathyroid hormone; hypnotics; immunosuppressants; muscle relaxants; parasympatholytics; parasympathomimetrics; prostaglandins; proteins; peptides; psychostimulants; sedatives; and tranquilizers.
- The beneficial agents used in the present invention include classical small molecular weight therapeutic agents commonly referred to as drugs including all classes of action as exemplified by, but not limited to: antiproliferatives, antithrombins, antiplatelet, antilipid, anti-inflammatory, angiogenic, anti-angiogenic, vitamins, ACE inhibitors, vasoactive substances, antimitotics, metello-proteinase inhibitors, NO donors, estradiols, anti-sclerosing agents, alone or in combination. Beneficial agent also includes larger molecular weight substances with drug like effects on target tissue sometimes called biologic agents including but not limited to: peptides, lipids, protein drugs, enzymes, oligonucleotides, ribozymes, genetic material, prions, virus, bacteria, and eucaryotic cells such as endothelial cells, monocyte/macrophages or vascular smooth muscle cells to name but a few examples. The therapeutic agent may also be a pro-drug, which metabolizes into the desired drug when administered to a host. Other beneficial agents may include but not be limited to physical agents such as microcapsules, microspheres, microbubbles, liposomes, niosomes, radioactive isotopes, emulsions, dispersions, or agents activated by some other form of energy such as light or ultrasonic energy, or by other circulating molecules that can be systemically administered.
- The embodiment of the invention shown in FIGS. 1 and 2 can be further refined by using Finite Element Analysis and other techniques to optimize the deployment of the beneficial agent within the openings of the struts and links. Basically, the shape and location of the
openings struts 18 with respect to the ductile hinges 20. - FIG. 3 illustrates a further preferred embodiment of the present invention, wherein like reference numerals have been used to indicate like components. The
tissue supporting device 100 includes a plurality ofcylindrical tubes 12 connected by S-shapedbridging elements 14. Each of thecylindrical tubes 12 has a plurality ofaxial slots 16 extending from an end surface of the cylindrical tube toward an opposite end surface. Formed between theslots 16 is a network ofaxial struts 18 and links 22. Eachindividual strut 18 is linked to the rest of the structure through a pair of ductile hinges 20, one at each end, which act as stress/strain concentration features. Each of the ductile hinges 20 is formed between anarc surface 28 and aconcave notch surface 29. - At intervals along the neutral axis of the
struts 18, at least one and more preferably a series ofopenings 24′ are formed by laser drilling or any other means known to one skilled in the art. Similarly, at least one and preferably a series ofopenings 26′ are formed at selected locations in thelinks 22. Although the use ofopenings 24′, 26′ in both thestruts 18 andlinks 22 is preferred, it should be clear to one skilled in the art that openings could be formed in only one of the struts and links. In the illustrated embodiment, theopenings 24′ in thestruts 18 are generally rectangular whereas theopenings 26′ in thelinks 22 are polygonal. It should be apparent to one skilled in the art, however, that openings of any geometrical shape or configuration could of course be used, and that the shape ofopenings openings openings 24′, 26′ may be loaded with an agent, most preferably a beneficial agent, for delivery to the vessel in which thetissue support device 100 is deployed. Although theopenings 24′, 26′ are preferably through openings, they may also be recesses extending only partially through the thickness of the struts and links. - The relatively large, protected
openings - The volume of beneficial agent that can be delivered using through openings is about 3 to 10 times greater than the volume of a 5 micron coating covering a stent with the same stent/vessel wall coverage ratio. This much larger beneficial agent capacity provides several advantages. The larger capacity can be used to deliver multi-drug combinations, each with independent release profiles, for improved efficacy. Also, larger capacity can be used to provide larger quantities of less aggressive drugs and to achieve clinical efficacy without the undesirable side-effects of more potent drugs, such as retarded healing of the endothelial layer.
- Through openings also decrease the surface area of the beneficial agent bearing compounds to which the vessel wall surface is exposed. For typical devices with beneficial agent openings, this exposure decreases by a factors ranging from about 6:1 to 8:1, by comparison with surface coated stents. This dramatically reduces the exposure of vessel wall tissue to polymer carriers and other agents that can cause inflammation, while simultaneously increasing the quantity of beneficial agent delivered, and improving control of release kinetics.
- FIG. 4 shows an enlarged view of one of the
struts 18 ofdevice 100 disposed between a pair of ductile hinges 20 having a plurality ofopenings 24′. FIG. 5 illustrates a cross section of one of theopenings 24′ shown in FIG. 4. FIG. 6 illustrates the same cross section when abeneficial agent 36 has been loaded into theopening 24′ of thestrut 18. Optionally, after loading theopening 24′ and/or theopening 26′ with abeneficial agent 36, the entire exterior surface of the stent can be coated with a thin layer of abeneficial agent 38, which may be the same as or different from thebeneficial agent 36, as schematically shown in FIG. 7. Still further, another variation of the present invention would coat the outwardly facing surfaces of the stent with a firstbeneficial agent 38 while coating the inwardly facing surfaces of the stent with a differentbeneficial agent 39, as illustrated in FIG. 8. The inwardly facing surface of the stent would be defined as at least the surface of the stent which, after expansion, forms the inner passage of the vessel. The outwardly facing surface of the stent would be defined as at least the surface of the stent which, after expansion, is in contact with and directly supports the inner wall of the vessel. Thebeneficial agent 39 coated on the inner surfaces may be a barrier layer which prevents thebeneficial agent 36 from passing into the lumen of the blood vessel and being washed away in the blood stream. - FIG. 9 shows a cross section of an
opening 24 in which one or more beneficial agents have been loaded into theopening 24 indiscrete layers 50. One method of creating such layers is to deliver a solution comprising beneficial agent, polymer carrier, and a solvent into the opening and evaporating the solvent to create a thin solid layer of beneficial agent in the carrier. Other methods of delivering the beneficial agent can also be used to create layers. According to another method for creating layers, a beneficial agent may be loaded into the openings alone if the agent is structurally viable without the need for a carrier. The process can then be repeated until each opening is partially or entirely filled. - In a typical embodiment, the total depth of the
opening 24 is about 125 to about 140 microns, and the typical layer thickness would be about 2 to about 50 microns, preferably about 12 microns. Each typical layer is thus individually about twice as thick as the typical coating applied to surface-coated stents. There would be at least two and preferably about ten to twelve such layers in a typical opening, with a total beneficial agent thickness about 25 to 28 times greater than a typical surface coating. According to one preferred embodiment of the present invention, the openings have an area of at least 5×10−6 square inches, and preferably at least 7×10−6 square inches. - Since each layer is created independently, individual chemical compositions and pharmacokinetic properties can be imparted to each layer. Numerous useful arrangements of such layers can be formed, some of which will be described below. Each of the layers may include one or more agents in the same or different proportions from layer to layer. The layers may be solid, porous, or filled with other drugs or excipients.
- FIG. 9 shows the simplest arrangement of layers including
identical layers 50 that together form a uniform, homogeneous distribution of beneficial agent. If the carrier polymer were comprised of a biodegradable material, then erosion of the beneficial agent containing carrier would occur on both faces of the opening at the same time, and beneficial agent would be released at an approximately linear rate over time corresponding to the erosion rate of the carrier. This linear or constant release rate is referred to as a zero order delivery profile. Use of biodegradable carriers in combination with through openings is especially useful, to guarantee 100% discharge of the beneficial agent within a desired time without creating virtual spaces or voids between the radially outermost surface of the stent and tissue of the vessel wall. When the biodegradable material in the through openings is removed, the openings may provide a communication between the strut-covered vessel wall and the blood stream. Such communication may accelerate vessel healing and allow the ingrowth of cells and extracellular components that more thoroughly lock the stent in contact with the vessel wall. Alternatively, some through-openings may be loaded with beneficial agent while others are left unloaded. The unloaded holes could provide an immediate nidus for the ingrowth of cells and extracellular components to lock the stent into place, while loaded openings dispense the beneficial agent. - The advantage of complete erosion using the through openings over surface coated stents opens up new possibilities for stent-based therapies. In the treatment of cardiac arrhythmias, such as atrial fibrillation both sustained and paroxysmal, sustained ventricular tachycardia, super ventricular tachycardia including reentrant and ectopic, and sinus tachycardia, a number of techniques under development attempt to ablate tissue in the pulmonary veins or some other critical location using various energy sources, e.g. microwaves, generally referred to as radio-frequency ablation, to create a barrier to the propagation of undesired electrical signals in the form of scar tissue. These techniques have proven difficult to control accurately. A stent based therapy using through openings, biodegradable carriers, and associated techniques described herein could be used to deliver a chemically ablative agent in a specific, precise pattern to a specific area for treatment of atrial fibrillation, while guaranteeing that none of the inherently cytotoxic ablating agent could be permanently trapped in contact with the tissue of the vessel wall.
- If, on the other hand, the goal of a particular therapy is to provide a long term effect, beneficial agents located in openings provide an equally dramatic advantage over surface coated devices. In this case, a composition comprising a beneficial agent and a non-biodegradable carrier would be loaded into the through openings, preferably in combination with a diffusion barrier layer as described below. To continue the cardiac arrhythmias example, it might be desirable to introduce a long-term anti-arrhythmic drug near the ostia of the pulmonary veins or some other critical location. The transient diffusion behavior of a beneficial agent through a non-biodegradable carrier matrix can be generally described by Fick's second law:

- Where C is the concentration of beneficial agent at cross section x, x is either the thickness of a surface coating or depth of a through opening, D is the diffusion coefficient and t is time. The solution of this partial differential equation for a through opening with a barrier layer will have the form of a normalized probability integral or Gaussian Error Function, the argument of which will contain
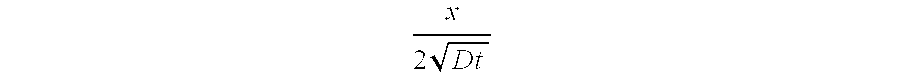
- the term
- To compare the time intervals over which a given level of therapy can be sustained for surface coatings vs. through openings, we can use Fick's Second Law to compare the times required to achieve equal concentrations at the most inward surfaces of the coating and opening respectively, i.e. the values of x and t for which the arguments

- of the Error Function are equal:
- The ratio of diffusion times to achieve comparable concentrations thus varies as the square of the ratio of depths. A typical opening depth is about 140 microns while a typical coating thickness is about 5 micron; the square of this ratio is 784, meaning that the effective duration of therapy for through openings is potentially almost three orders of magnitude greater for through openings than for surface coatings of the same composition. The inherent non-linearity of such release profiles can in part be compensated for in the case of through openings, but not in thin surface coatings, by varying the beneficial agent concentration of layers in a through opening as described below. It will be recalled that, in addition to this great advantage in beneficial agent delivery duration, through openings are capable of delivering a 3 to 10 times greater quantity of beneficial agent, providing a decisive overall advantage in sustained therapies. The diffusion example above illustrates the general relationship between depth and diffusion time that is characteristic of a wider class of solid state transport mechanisms.
- Beneficial agent that is released to the radially innermost or inwardly facing surface known as the lumen facing surface of an expanded device may be rapidly carried away from the targeted area, for example by the bloodstream, and thus lost. Up to half of the total agent loaded in such situations may have no therapeutic effect due to being carried away by the bloodstream. This is probably the case for all surface coated stents as well as the through opening device of FIG. 9.
- FIG. 10 shows a device in which the
first layer 52 is loaded into a throughopening 24 such that the inner surface of the layer is substantially co-planar with the inwardly facingsurface 54 of the cylindrical device. Thefirst layer 52 is comprised of a material called a barrier material which blocks or retards biodegradation of subsequent layers in the inwardly facing direction toward the vessel lumen, and/or blocks or retards diffusion of the beneficial agent in that direction. Biodegradation of other layers or beneficial agent diffusion can then proceed only in the direction of the outwardly facingsurface 56 of the device, which is in direct contact with the targeted tissue of the vessel wall. Thebarrier layer 52 may also function to prevent hydration of inner layers of beneficial agent and thus prevent swelling of the inner layers when such layers are formed of hygroscopic materials. Thebarrier layer 52 may further be comprised of a biodegradable material that degrades at a much slower rate than the biodegradable material in the other layers, so that the opening will eventually be entirely cleared. Providing abarrier layer 52 in the most inwardly facing surface of a through-opening thus guarantees that the entire load of beneficial agent is delivered to the target area in the vessel wall. It should be noted that providing a barrier layer on the inwardly facing surface of a surface-coated stent without openings does not have the same effect; since the beneficial agent in such a coating cannot migrate through the metal stent to the target area on the outer surface, it simply remains trapped on the inner diameter of the device, again having no therapeutic effect. - Barrier layers can be used to control beneficial agent release kinetics in more sophisticated ways. A
barrier layer 52 with a pre-determined degradation time could be used to deliberately terminate the beneficial agent therapy at a pre-determined time, by exposing the underlying layers to more rapid bio-degradation from both sides. Barrier layers can also be formulated to be activated by a separate, systemically applied agent. Such systemically applied agent could change the porosity of the barrier layer and/or change the rate of bio-degradation of the barrier layer or the bulk beneficial agent carrier. In each case, release of the beneficial agent could be activated by the physician at will by delivery of the systemically applied agent. A further embodiment of physician activated therapy would utilize a beneficial agent encapsulated in micro-bubbles and loaded into device openings. Application of ultrasonic energy from an exterior of the body could be used to collapse the bubbles at a desired time, releasing the beneficial agent to diffuse to the outwardly facing surface of the reservoirs. These activation techniques can be used in conjunction with the release kinetics control techniques described herein to achieve a desired drug release profile that can be activated and/or terminated at selectable points in time. - FIG. 11A shows an arrangement of layers provided in a through opening in which layers 50 of a beneficial agent in a biodegradable carrier material, are alternated with
layers 58 of the biodegradable carrier material alone, with no active agent loaded, and abarrier layer 52 is provided at the inwardly facing surface. As shown in the release kinetics plot of FIG. 11B, such an arrangement releases beneficial agent in three programmable bursts or waves achieving a stepped or pulsatile delivery profile. The use of carrier material layers without active agent creates the potential for synchronization of drug release with cellular biochemical processes for enhanced efficacy. - Alternatively, different layers could be comprised of different beneficial agents altogether, creating the ability to release different beneficial agents at different points in time, as shown in FIG. 12. For example, in FIG. 12, a
layer 60 of anti-thrombotic agent could be deposited at the inwardly facing surface of the stent, followed by abarrier layer 52 and alternating layers ofanti-proliferatives 62 andanti-inflamatories 64. This configuration could provide an initial release of anti-thrombotic agent into the bloodstream while simultaneously providing a gradual release of anti-proliferatives interspersed with programmed bursts of anti-inflammatory agents to the vessel wall. The configurations of these layers can be designed to achieve the agent delivery bursts at particular points in time coordinated with the body's various natural healing processes. - A further alternative is illustrated in FIG. 13. Here the concentration of the same beneficial agent is varied from layer to layer, creating the ability to generate release profiles of arbitrary shape. Progressively increasing the concentration of agent in the
layers 66 with increasing distance from the outwardly facingsurface 56, for example, produces a release profile with a progressively increasing release rate, which would be impossible to produce in a thin surface coating. - Another general method for controlling beneficial agent release kinetics is to alter the beneficial agent flux by changing the surface area of drug elution sources as a function of time. This follows from Fick's First Law, which states that the instantaneous molecular flux is proportional to surface area, among other factors:

- Where ∂N/∂t is the number of molecules per unit time, A is the instantaneous drug eluting surface area, D is the diffusivity, and C is the concentration. The drug eluting surface area of a surface coated stent is simply the surface area of the stent itself. Since this area is fixed, this method of controlling release kinetics is not available to surface coated devices. Through openings, however, present several possibilities for varying surface area as a function of time.
- In the embodiment of FIG. 14, beneficial agent is provided in the
openings 24 in the form of microspheres, particles or the like.Individual layers 70 can then be created that contain these particles. Further, the particle size can be varied from layer to layer. For a given layer volume, smaller particle sizes increase the total particle surface area in that layer, which has the effect of varying the total surface area of the beneficial agent from layer to layer. Since the flux of drug molecules is proportional to surface area, the total drug flux can be adjusted from layer to layer by changing the particle size, and the net effect is control of release kinetics by varying particle sizes within layers. - A second general method for varying drug eluting surface area as a function of time is to change the shape or cross-sectional area of the drug-bearing element along the axis of the opening. FIG. 15A shows an
opening 70 having a conical shape cut into the material of the stent itself. Theopening 70 may then be filled withbeneficial agent 72 in layers as described above or in another manner. In this embodiment, abarrier layer 74 may be provided on the inwardly facing side of theopening 70 to prevent thebeneficial agent 72 from passing into the blood stream. In this example, the drug eluting surface area At would continuously diminish (from FIG. 15A to FIG. 15B) as the bio-degradable carrier material erodes, yielding the elution pattern of FIG. 15C. - FIG. 16A shows a simple cylindrical through-opening 80 in which a preformed,
inverted cone 82 of beneficial agent has been inserted. The rest of the throughopening 80 is then back-filled with abiodegradable substance 84 with a much slower rate of degradation or a non-biodegradable substance, and the inwardly facing opening of the through opening is sealed with abarrier layer 86. This technique yields the opposite behavior to the previous example. The drug-eluting surface area At continuously increases with time between FIGS. 16A and 16B, yielding the elution pattern of FIG. 16C. - The changing
cross section openings 70 of FIG. 15A and the non-biodegradable backfilling techniques of FIG. 16A may be combined with any of the layered agent embodiments of FIGS. 9-14 to achieve desired release profiles. For example, the embodiment of FIG. 15A may use the varying agent concentration layers of FIG. 13 to more accurately tailor a release curve to a desired profile. - The process of preforming the beneficial agent plug 82 to a special shape, inserting in a through opening, and back-filling with a second material can yield more complex release kinetics as well. FIG. 17A shows a through
opening 90 in which a sphericalbeneficial agent plug 92 has been inserted. The resulting biodegradation of the sphere, in which the cross sectional surface area varies as a sinusoidal function of depth, produces a flux density which is roughly a sinusoidal function of time, FIG. 17B. Other results are of course possible with other profiles, but none of these more complex behaviors could be generated in a thin, fixed-area surface coating. - An alternative embodiment of FIGS. 18A-18C use a
barrier layer 52′ with anopening 96 to achieve the increasing agent release profile of FIG. 16C. As shown in FIG. 18A, theopening 24 is provided with aninner barrier layer 52 and multiple beneficial agent layers 50 as in the embodiment of FIG. 10. An additionalouter barrier layer 52′ is provided with asmall hole 96 for delivery of the agent to the vessel wall. As shown in FIQS. 18B and 18C, the beneficialagent containing layers 50 degrade in a hemispherical pattern resulting in increasing surface area for agent delivery over time and thus, an increasing agent release profile. - FIG. 19 illustrates an alternative embodiment in which an opening in the tissue supporting device is loaded with cylindrical layers of beneficial agent. According to one method of forming the device of FIG. 19, the entire device is coated with
sequential layers interior surface 54 andexterior surface 56 of the device are then stripped to remove the beneficial agent on these surfaces leaving the cylindrical layers of beneficial agent in the openings. In this embodiment, a central opening remains after the coating layers have been deposited which allows communication between theouter surface 56 andinner surface 54 of the tissue supporting device. - In the embodiment of FIG. 19, the cylindrical layers are eroded sequentially. This can be used for pulsatile delivery of different beneficial agents, delivery of different concentrations of beneficial agents, or delivery of the same agent. As shown in FIG. 19, the ends of the
cylindrical layers - FIG. 20 illustrates a further alternative embodiment of the invention in which different beneficial agents are positioned in different holes of an expandable
medical device 300. A first beneficial agent is provided inholes 330 a at the ends of the device and a second beneficial agent is provided inholes 330 b at a central portion of the device. The beneficial agent may contain different drugs, the same drugs in different concentrations, or different variations of the same drug. The embodiment of FIG. 20 may be used to provide an expandablemedical device 300 with either “hot ends” or “cool ends.” - Preferably, each end portion of the
device 300 which includes theholes 330 a containing the first beneficial agent extends at least one hole and up to about 15 holes from the edge. This distance corresponds to about 0.005 to about 0.1 inches from the edge of an unexpanded device. The distance from the edge of thedevice 300 which includes the first beneficial agent is preferably about one section, where a section is defined between the bridging elements. - Different beneficial agents containing different drugs may be disposed in different openings in the stent. This allows the delivery of two or more beneficial agents from a single stent in any desired delivery pattern. Alternatively, different beneficial agents containing the same drug in different concentrations may be disposed in different openings. This allows the drug to be uniformly distributed to the tissue with a non-uniform device structure.
- The two or more different beneficial agents provided in the devices described herein may contain (1) different drugs; (2) different concentrations of the same drug; (3) the same drug with different release kinetics, i.e., different matrix erosion rates; or (4) different forms of the same drug. Examples of different beneficial agents formulated containing the same drug with different release kinetics may use different carriers to achieve the elution profiles of different shapes. Some examples of different forms of the same drug include forms of a drug having varying hydrophilicity or lipophilicity.
- In one example of the
device 300 of FIG. 20, theholes 330 a at the ends of the device are loaded with a first beneficial agent comprising a drug with a high lipophilicity whileholes 330 b at a central portion of the device are loaded with a second beneficial agent comprising the drug with a lower lipophilicity. The first high lipophilicity beneficial agent at the “hot ends” will diffuse more readily into the surrounding tissue reducing the edge effect restenosis. - The
device 300 may have an abrupt transition line at which the beneficial agent changes from a first agent to a second agent. For example, all openings within 0.05 inches of the end of the device may contain the first agent while the remaining openings contain the second agent. Alternatively, the device may have a gradual transition between the first agent and the second agent. For example, a concentration of the drug in the openings can progressively increase (or decrease) toward the ends of the device. In another example, an amount of a first drug in the openings increases while an amount of a second drug in the openings decreases moving toward the ends of the device. - FIG. 21 illustrates a further alternative embodiment of an expandable
medical device 400 in which different beneficial agents are positioned indifferent openings - In addition to the use of different beneficial agents in different openings to achieve different drug concentrations at different defined areas of tissue, the loading of different beneficial agents in different openings may be used to provide a more even spatial distribution of the beneficial agent delivered in instances where the expandable medical device has a non-uniform distribution of openings in the expanded configuration.
- For example, in many of the known expandable devices and for the device illustrated in FIG. 22 the coverage of the
device 500 is greater at thecylindrical tube portions 512 of the device than at the bridgingelements 514. Coverage is defined as the ratio of the device surface area to the area of the lumen in which the device is deployed. When a device with varying coverage is used to deliver a beneficial agent contained in openings in the device, the beneficial agent concentration delivered to the tissue adjacent thecylindrical tube portions 512 is greater that the beneficial agent delivered to the tissue adjacent the bridgingelements 514. In order to address this longitudinal variation in device structure and other variations in device coverage which lead to uneven beneficial agent delivery concentrations, the concentration of the beneficial agent may be varied in the openings at portions of the device to achieve a more even distribution of the beneficial agent throughout the tissue. In the case of the embodiment of FIG. 22, theopenings 530 a in thetube portions 512 include a beneficial agent with a lower drug concentration than theopenings 530 b in the bridgingelements 514. The uniformity of agent delivery may be achieved in a variety of manners including varying the drug concentration, the opening diameter or shape, the amount of agent in the opening (i.e., the percentage of the opening filed), the matrix material, or the form of the drug. - In addition to the delivery of different beneficial agents to the mural side of the expandable medical device for treatment of the vessel wall, beneficial agents may be delivered to the lumenal side of the expandable medical device. Drugs which are delivered into the blood stream from the lumenal side of the device can be located at a proximal end of the device or a distal end of the device.
- The methods for loading different beneficial agents into different openings in an expandable medical device may include known techniques such as dipping and coating and also known piezoelectric micro-jetting techniques. Micro-injection devices may be computer controlled to deliver precise amounts of two or more liquid beneficial agents to precise locations on the expandable medical device in a known manner. For example, a dual agent jetting device may deliver two agents simultaneously or sequentially into the openings. When the beneficial agents are loaded into through openings in the expandable medical device, a lumenal side of the through openings may be blocked during loading by a resilient mandrel allowing the beneficial agents to be delivered in liquid form, such as with a solvent. The beneficial agents may also be loaded by manual injection devices.
- Therapeutic Layer Formulations
- The therapeutic agent layers of the present invention are beneficial agents comprised of a matrix and at least one therapeutic agent. The term “matrix” or “biocompatible matrix” are used interchangeably to refer to a medium or material that, upon implantation in a subject, does not elicit a detrimental response sufficient to result in the rejection of the matrix. The matrix typically does not provide any therapeutic responses itself, though the matrix may contain or surround a therapeutic agent, a therapeutic agent, an activating agent or a deactivating agent, as defined herein. A matrix is also a medium that may simply provide support, structural integrity or structural barriers. The matrix may be polymeric, non-polymeric, hydrophobic, hydrophilic, lipophilic, amphiphilic, and the like.
- The matrix of the therapeutic agent layers can be made from pharmaceutically acceptable polymers, such as those typically used in medical devices. The term “polymer” refers to molecules formed from the chemical union of two or more repeating units, called monomers. Accordingly, included within the term “polymer” may be, for example, dimers, trimers and oligomers. The polymer may be synthetic, naturally-occurring or semisynthetic. In preferred form, the term “polymer” refers to molecules which typically have a M w greater than about 3000 and preferably greater than about 10,000 and a Mw that is less than about 10 million, preferably less than about a million and more preferably less than about 200,000. Examples of polymers include but are not limited to, poly-α-hydroxy acid esters such as, polylactic acid, polyglycolic acid, polylactic-co-glycolic acid, polylactic acid-co-caprolactone; polyethylene glycol and polyethylene oxide; polyvinyl pyrrolidone; polyorthoesters; polysaccharides and polysaccharide derivatives such as polyhyaluronic acid, polyalginic acid, chitin, chitosan, cellulose, hydroxyehtylcellulose, hydroxypropylcellulose, carboxymethylcellulose; polypeptides, and proteins such as polylysine, polyglutamic acid, albumin; polyanhydrides; polyhydroxy alkonoates such as polyhydroxy valerate, polyhydroxy butyrate, and the like, and copolymers thereof. The polymers and copolymers can be prepared by methods well known in the art (see, for example, Rempp and Merril: Polymer Synthesis, 1998, John Wiley and Sons) in or can be used as purchased from Alkermes, in Cambridge, Mass. or Birmingham Polymer Inc., in Birmingham, Ala.
- The preferred co-polymer for use in the present invention are poly(lactide-co-glycolide) (PLGA) polymers. The rate at which the polymer erodes is determined by the selection of the ratio of lactide to glycolide within the copolymer, the molecular weight of each polymer used, and the crystallinity of the polymers used.
- Bioerodible polymers may also be used to form barrier layers that erode at a rate that can be predetermined base on the composition and that contain no therapeutic agent.
- Additives in Protective, Barrier, or Therapeutic Layer Formulations
- Typical additives that may be included in a bioerodible matrix are well known to those skilled in the art (see Remington: The Science and Practice of Pharmacy, Gennaro, ed., Mack Publishing Co., Easton, Pa., 19th ed., 1995) and include but are not limited to pharmaceutically acceptable excipients, adjuvants, carriers, antioxidants, preservatives, buffers, antacids, emulsifiers, inert fillers, fragrances, thickeners, tackifiers, opacifiers, gelling agents, stabilizers, surfactants, emolliehts, coloring agents, and the like.
- Typical formulations for therapeutic agents incorporated in these medical devices are well known to those skilled in the art and include but are not limited to solid particle dispersions, encapsulated agent dispersions, and emulsions, suspensions, liposomes or microparticles, wherein said liposome or microparticle comprise a homogeneous or heterogeneous mixture of the therapeutic agent.
- The term “homogeneously disposed” refers to a component which is mixed uniformly in a matrix in such a manner that the component is macroscopically indistinguishable from the matrix itself. An example of a homogeneously disposed component is a drug formulation such as a microemulsion in which small beads of oil are dispersed uniformly in water.
- The term “heterogeneously disposed” refers to a component which is mixed non-uniformly into a matrix in such a manner that the component is macroscopically distinguishable from the matrix itself. An example of a heterogeneously disposed component is a simple emulsion in which the beads of oil in the water are large enough to cause a turbidity to the solution and can be seen settling out of solution over time. Heterogeneously disposed compositions also include encapsulated formulations where a component, such as a protective layer, is layered onto or around a therapeutic agent or a therapeutic layer, forming a protective shell.
- The amount of the drug that is present in the device, and that is required to achieve a therapeutic effect, depends on many factors, such as the minimum necessary dosage of the particular drug, the condition to be treated, the chosen location of the inserted device, the actual compound administered, the age, weight, and response of the individual patient, the severity of the patient's symptoms, and the like.
- The appropriate dosage level of the therapeutic agent, for more traditional routes of administration, are known to one skilled in the art. These conventional dosage levels correspond to the upper range of dosage levels for compositions, including a physiologically active substance and traditional penetration enhancer. However, because the delivery of the active substance occurs at the site where the drug is required, dosage levels significantly lower than a conventional dosage level may be used with success. Ultimately, the percentage of therapeutic agent in the composition is determined by the required effective dosage, the therapeutic activity of the particular formulation, and the desired release profile. In general, the active substance will be present in the composition in an amount from about 0.0001% to about 99%, more preferably about 0.01% to about 80% by weight of the total composition depending upon the particular substance employed. However, generally the amount will range from about 0.01% to about 75% by weight of the total composition, with levels of from about 25% to about 75% being preferred.
- Protective and Barrier Layer Formulations
- The protective and barrier layers of the present invention are beneficial agents comprised of a bioerodible matrix and optionally contain additional additives, therapeutic agents, activating agents, deactivating agents, and the like. Either a property of the chosen material of the protective or barrier layer, or a chemical embedded in the protective or barrier layer provides protection from deactivating processes or conditions for at least one therapeutic agent. In addition to the polymer materials described above, the protective or barrier layer may also be comprised of pharmaceutically acceptable lipids or lipid derivatives, which are well known in the art and include but are not limited to fatty acids, fatty acid esters, lysolipids, phosphocholines, (Avanti Polar Lipids, Alabaster, Ala.), including 1-alkyl-2-acetoyl-sn-glycero 3-phosphocholines, and 1-alkyl-2-hydroxy-sn-glycero 3-phosphocholines; phosphatidylcholine with both saturated and unsaturated lipids, including dioleoylphosphatidylcholine; dimyristoyl-phosphatidylcholine; dipentadecanoylphosphatidylcholine; dilauroylphosphatidyl-choline; dipalmitoylphosphatidylcholine (DPPC); distearoylphosphatidylcholine (DSPC); and diarachidonylphosphatidylcholine (DAPC); phosphatidyl-ethanolamines, such as dioleoylphosphatidylethanolamine, dipahnitoyl-phosphatidylethanolamine (DPPE) and distearoylphosphatidylefhanolamine (D SPE); phosphatidylserine; phosphatidylglycerols, including distearoylphosphatidylglycerol (DSPG); phosphatidylinositol; sphingolipids such as sphingomyelin; glucolipids; sulfatides; glycosphingolipids; phosphatidic acids, such as dipahmitoylphosphatidic acid (DPPA) and distearoylphosphatidic acid (DSPA); palmitic acid; stearic acid; arachidonic acid; oleic acid; lipids bearing polymers, such as chitin, hyaluronic acid, polyvinylpyrrolidone or polyethylene glycol (PEG), also referred to herein as “pegylated lipids”, with preferred lipids bearing polymers including DPPE-PEG (DPPE-PEG), which refers to the lipid DPPE having a PEG polymer attached thereto, including, for example, DPPE-PEG5000, which refers to DPPE having attached thereto a PEG polymer having a mean average molecular weight of about 5000; lipids bearing sulfonated mono-, di-, oligo- or polysaccharides; cholesterol, cholesterol sulfate and cholesterol hemisuccinate; tocopherol hemisuccinate; lipids with ether and ester-linked fatty acids; polymerized lipids (a wide variety of which are well known in the art); diacetyl phosphate; dicetyl phosphate; stearylamine; cardiolipin; phospholipids with short chain fatty acids of about 6 to about 8 carbons in length; synthetic phospholipids with asymmetric acyl chains, such as, for example, one acyl chain of about 6 carbons and another acyl chain of about 12 carbons; ceramides; non-ionic liposomes including niosomes such as polyoxyethylene fatty acid esters, polyoxyethylene fatty alcohols, polyoxyethylene fatty alcohol ethers, polyoxyethylated sorbitan fatty acid esters, glycerol polyethylene glycol oxystearate, glycerol polyethylene glycol ricinoleate, ethoxylated soybean sterols, ethoxylated castor oil, polyoxyethylene-polyoxypropylene polymers, and polyoxyethylene fatty acid stearates; sterol aliphatic acid esters including cholesterol sulfate, cholesterol butyrate, cholesterol iso-butyrate, cholesterol palmitate, cholesterol stearate, lanosterol acetate, ergosterol palmitate, and phytosterol n-butyrate; sterol esters of sugar acids including cholesterol glucuronide, lanosterol glucuronide, 7-dehydrocholesterol glucuronide, ergosterol glucuronide, cholesterol gluconate, lanosterol gluconate, and ergosterol gluconate; esters of sugar acids and alcohols including lauryl glucuronide, stearoyl glucuronide, myristoyl glucuronide, lauryl gluconate, myristoyl gluconate, and stearoyl gluconate; esters of sugars and aliphatic acids including sucrose acetate isobutyrate (SAIB), sucrose laurate, fructose laurate, sucrose palritate, sucrose stearate, glucuronic acid, gluconic acid and polyuronic acid; saponins including sarsasapogenin, smilagenin, hederagenin, oleanolic acid, and digitoxigenin; glycerol dilaurate, glycerol trilaurate, glycerol monolaurate, glycerol dipalmitate, glycerol and glycerol esters including glycerol tripalmitate, glycerol monopalmitate, glycerol distearate, glycerol tristearate, glycerol monostearate, glycerol monomyristate, glycerol dimyristate, glycerol trimyristate; long chain alcohols including n-decyl alcohol, lauryl alcohol, myristyl alcohol, cetyl alcohol, and n-octadecyl alcohol; 1,2-dioleoyl-sn-glycerol; 1,2-dipalmitoyl-sn-3-succinylglycerol; 1,3-dipalmitoyl-2-succinylglycerol; 1-hexadecyl-2-palmitoylglycerophosphoethanolamine and palmitoylhomocysteine, and/or combinations thereof.
- If desired, a cationic lipid may be used, such as, for example, N-[1-(2,3-dioleoyloxy)propyl]-N,N,N-trimethylammonium chloride (DOTMA), 1,2-dioleoyloxy-3-(trimethylammonio)propane (DOTAP); and 1,2-dioleoyl-3-(4′-trimethylammonio)butanoyl-sn-glycerol (DOTB). If a cationic lipid is employed in the lipid compositions, the molar ratio of cationic lipid to non-cationic lipid may be, for example, from about 1:1000 to about 1:100. Preferably, the molar ratio of cationic lipid to non-cationic lipid may be from about 1:2 to about 1:10, with a ratio of from about 1:1 to about 1:2.5 being preferred. Even more preferably, the molar ratio of cationic lipid to non-cationic lipid may be about 1:1.
- These lipid materials are well known in the art and can be used as purchased from Avanti, Burnaby, B.C. Canada.
- The preferred lipids for use in the present invention are phosphatidyl-choline, phosphatidylethanolamine, phosphatidylserine, sphingomyelin as well as synthetic phospholipids such as dimyristoyl phosphatidylcholine, dipalmitoyl phosphatidylcholine, distearoyl phosphatidylcholine, distearoyl phosphatidyl-glycerol, dipalmitoyl phosphatidylglycerol, dimyristoyl phosphatidylserine, distearoyl phosphatidylserine and dipalmitoyl phosphatidylserine.
- The rate at which the bioerodible matrix erodes is determined by the choice of lipid, the molecular weight, and the ratio of the chosen materials.
- The protective or barrier layer can erode by either chemical or physical erosion mechanisms. If the layer erodes by a physical mechanism, the layer is typically a thin film from about 0.1 μm to about 3 μm of a non-polymeric material embedded between two polymeric layers. In this instance, the structural integrity of the protective or barrier layer is maintained by the presence of both of these polymeric layers. When the polymeric material closest to the luminal surface erodes away, the protective or barrier layer breaks apart by the physical forces exerted on it from the remaining polymeric layer. In another embodiment, the protective or barrier layer is eroded by chemical interactions, dissolution in water, hydrolysis, or reaction with enzymes.
- One function of the protective or barrier layer is to protect one or more therapeutic agents from deactivating or degrading conditions. The protection may come from the properties of the material when, for example, a hydrophobic protective or barrier layer would protect a water sensitive agent from water by resisting the influx of moisture. The protective or barrier layer may also act as a physical barrier. For example, a protective or barrier layer comprised of a hydrogel may allow water to be absorbed by the gel, and allow any agents contained within the gel to diffuse out of the gel into the reaction environment. The hydrogel, however, would prevent enzymes from penetrating the layer, thereby protecting any agents contained within from the enzyme. The term “hydrogel” refers to cross-linked polymeric material in which the liquid component is water. Hydrogels may be prepared by cross-linking certain polymers and lipids disclosed herein.
- Finally the protective or barrier layer does not have to act as a barrier. The protective or barrier layer may protect a therapeutic agent by releasing an agent, such as an activating agent or a deactivating agent, into the reaction environment prior to the release of the therapeutic agent.
- A therapeutic agent may be incorporated directly in the protective or barrier layer. The therapeutic agent can be heterogeneously or homogeneously dispersed in the protective or barrier layer. The therapeutic agent can be a drug, or a drug formulated into a microcapsule, niosome, liposome, microbubble, microsphere, or the like. In addition, the protective or barrier layer may contain more than one therapeutic agent. For example, a water sensitive drugs, such as a limus, or any other drug that must be administered through intravenous, intramuscular, or subcutaneously, could be incorporated in a hydrophobic matrix such as SAIB, or fatty acid ester.
- A therapeutic agent may also be disposed in a therapeutic agent layer, separate from the protective or barrier layer. In this case the protective or barrier layer may be adjacent to the therapeutic agent layer and may serve to prevent or retard processes that would degrade or deactivate the therapeutic agent until the protective or barrier layer has substantially eroded. In this instance the protective or barrier layer is a barrier between a therapeutic layer and the reaction environment. This barrier may be a hydrophobic barrier that resists water absorption. The hydrophobic barrier would be used in conjunction with water-sensitive drugs as described above. Alternatively, the protective or barrier layer maybe a hydrogel that resists the absorbance of enzymes. An enzyme resistant barrier would used to protect an drug such as a DNA, RNA, peptide or protein based therapeutic agent.
- The protective or barrier layer may optionally include activating and deactivating agents for the purpose of preparing the reaction environment for the subsequent release of a therapeutic agent. These activating and deactivating agents are well known to those skilled in the art and include but are not limited to antacids, buffers, enzyme inhibitors, hydrophobic additives, and adjuvants. For example, Mg(OH) 2 in particles of about 0.5 μm to about 5 μm more preferably, about 1 μm incorporated in a PLGA polymer layer could be used in conjunction with any acid senstive drug. An example of an activating agent is chymotrypsin, which may be incorporated in polyvinyl pyrrolidone layer. The chymotrypsin, could be used to convert a pro-drug to an active drug.
- While the invention has been described in detail with reference to the preferred embodiments thereof, it will be apparent to one skilled in the art that various changes and modifications can be made and equivalents employed, without departing from the present invention.
Claims (29)
Priority Applications (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US10/857,201 US20040254635A1 (en) | 1998-03-30 | 2004-05-27 | Expandable medical device for delivery of beneficial agent |
| US11/925,344 US8361537B2 (en) | 1998-03-30 | 2007-10-26 | Expandable medical device with beneficial agent concentration gradient |
| US12/413,727 US20090228095A1 (en) | 1998-03-30 | 2009-03-30 | Expandable medical device for delivery of beneficial agent |
Applications Claiming Priority (9)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US7988198P | 1998-03-30 | 1998-03-30 | |
| US09/183,555 US6241762B1 (en) | 1998-03-30 | 1998-10-29 | Expandable medical device with ductile hinges |
| US68809200A | 2000-10-16 | 2000-10-16 | |
| US31425901P | 2001-08-20 | 2001-08-20 | |
| US09/948,989 US7208010B2 (en) | 2000-10-16 | 2001-09-07 | Expandable medical device for delivery of beneficial agent |
| US41248902P | 2002-09-20 | 2002-09-20 | |
| US10/253,020 US7208011B2 (en) | 2001-08-20 | 2002-09-23 | Implantable medical device with drug filled holes |
| US10/668,430 US20040127977A1 (en) | 2002-09-20 | 2003-09-22 | Expandable medical device with openings for delivery of multiple beneficial agents |
| US10/857,201 US20040254635A1 (en) | 1998-03-30 | 2004-05-27 | Expandable medical device for delivery of beneficial agent |
Related Parent Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US10/253,020 Continuation-In-Part US7208011B2 (en) | 1998-03-30 | 2002-09-23 | Implantable medical device with drug filled holes |
| US10/668,430 Continuation-In-Part US20040127977A1 (en) | 1998-03-30 | 2003-09-22 | Expandable medical device with openings for delivery of multiple beneficial agents |
Related Child Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US11/925,344 Continuation US8361537B2 (en) | 1998-03-30 | 2007-10-26 | Expandable medical device with beneficial agent concentration gradient |
| US12/413,727 Continuation US20090228095A1 (en) | 1998-03-30 | 2009-03-30 | Expandable medical device for delivery of beneficial agent |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| US20040254635A1 true US20040254635A1 (en) | 2004-12-16 |
Family
ID=46301357
Family Applications (3)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US10/857,201 Abandoned US20040254635A1 (en) | 1998-03-30 | 2004-05-27 | Expandable medical device for delivery of beneficial agent |
| US11/925,344 Expired - Fee Related US8361537B2 (en) | 1998-03-30 | 2007-10-26 | Expandable medical device with beneficial agent concentration gradient |
| US12/413,727 Abandoned US20090228095A1 (en) | 1998-03-30 | 2009-03-30 | Expandable medical device for delivery of beneficial agent |
Family Applications After (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US11/925,344 Expired - Fee Related US8361537B2 (en) | 1998-03-30 | 2007-10-26 | Expandable medical device with beneficial agent concentration gradient |
| US12/413,727 Abandoned US20090228095A1 (en) | 1998-03-30 | 2009-03-30 | Expandable medical device for delivery of beneficial agent |
Country Status (1)
| Country | Link |
|---|---|
| US (3) | US20040254635A1 (en) |
Cited By (70)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20050100582A1 (en) * | 2003-11-06 | 2005-05-12 | Stenzel Eric B. | Method and apparatus for controlled delivery of active substance |
| US7070590B1 (en) | 1996-07-02 | 2006-07-04 | Massachusetts Institute Of Technology | Microchip drug delivery devices |
| US20070055352A1 (en) * | 2005-09-07 | 2007-03-08 | Wendy Naimark | Stent with pockets for containing a therapeutic agent |
| WO2007108916A2 (en) * | 2006-03-15 | 2007-09-27 | Boston Scientific Scimed, Inc. | Drug delivery composition and methods of making same using nanofabrication |
| EP1858442A2 (en) * | 2005-03-14 | 2007-11-28 | Conor Medsystems, Inc. | Expandable medical device with openings for delivery of multiple beneficial agents |
| EP1890648A2 (en) * | 2005-06-06 | 2008-02-27 | Innovational Holdings, LLC | Implantable medical device with openings for delivery of beneficial agents with combination release kinetics |
| US20080243241A1 (en) * | 2007-03-28 | 2008-10-02 | Zhao Jonathon Z | Short term sustained drug-delivery system for implantable medical devices and method of making the same |
| WO2009014692A1 (en) * | 2007-07-24 | 2009-01-29 | Boston Scientific Limited | Stents with polymer-free coatings for delivering a therapeutic agent |
| WO2009055720A1 (en) | 2007-10-26 | 2009-04-30 | University Of Virginia Patent Foundation | System for treatment and imaging using ultrasonic energy and microbubbles and related method thereof |
| US20090163991A1 (en) * | 2007-12-19 | 2009-06-25 | Boston Scientific Scimed, Inc. | Stent |
| US20090274740A1 (en) * | 2008-05-01 | 2009-11-05 | Boston Scientific Scimed, Inc. | Drug-loaded medical devices and methods for manufacturing drug-loaded medical devices |
| US20090319032A1 (en) * | 2008-06-18 | 2009-12-24 | Boston Scientific Scimed, Inc | Endoprosthesis coating |
| US20100010621A1 (en) * | 2008-07-11 | 2010-01-14 | Biotronik Vi Patent Ag | Stent having biodegradable stent struts and drug depots |
| US20100028403A1 (en) * | 2008-07-31 | 2010-02-04 | Boston Scientific Scimed, Inc. | Medical devices for therapeutic agent delivery |
| US7842083B2 (en) | 2001-08-20 | 2010-11-30 | Innovational Holdings, Llc. | Expandable medical device with improved spatial distribution |
| US20110017346A1 (en) * | 2002-09-20 | 2011-01-27 | Innovational Holdings, Llc | Method and apparatus for loading a beneficial agent into an expandable medical device |
| US7931683B2 (en) | 2007-07-27 | 2011-04-26 | Boston Scientific Scimed, Inc. | Articles having ceramic coated surfaces |
| US7938855B2 (en) | 2007-11-02 | 2011-05-10 | Boston Scientific Scimed, Inc. | Deformable underlayer for stent |
| US7942926B2 (en) | 2007-07-11 | 2011-05-17 | Boston Scientific Scimed, Inc. | Endoprosthesis coating |
| US7951392B2 (en) | 2002-08-16 | 2011-05-31 | Boston Scientific Scimed, Inc. | Microarray drug delivery coatings |
| US20110143014A1 (en) * | 2009-12-11 | 2011-06-16 | John Stankus | Coatings with tunable molecular architecture for drug-coated balloon |
| US7976915B2 (en) | 2007-05-23 | 2011-07-12 | Boston Scientific Scimed, Inc. | Endoprosthesis with select ceramic morphology |
| US7981150B2 (en) | 2006-11-09 | 2011-07-19 | Boston Scientific Scimed, Inc. | Endoprosthesis with coatings |
| US7985252B2 (en) | 2008-07-30 | 2011-07-26 | Boston Scientific Scimed, Inc. | Bioerodible endoprosthesis |
| US7998192B2 (en) | 2008-05-09 | 2011-08-16 | Boston Scientific Scimed, Inc. | Endoprostheses |
| US8002821B2 (en) | 2006-09-18 | 2011-08-23 | Boston Scientific Scimed, Inc. | Bioerodible metallic ENDOPROSTHESES |
| US8002823B2 (en) | 2007-07-11 | 2011-08-23 | Boston Scientific Scimed, Inc. | Endoprosthesis coating |
| US8029554B2 (en) | 2007-11-02 | 2011-10-04 | Boston Scientific Scimed, Inc. | Stent with embedded material |
| US8048150B2 (en) | 2006-04-12 | 2011-11-01 | Boston Scientific Scimed, Inc. | Endoprosthesis having a fiber meshwork disposed thereon |
| US8052744B2 (en) | 2006-09-15 | 2011-11-08 | Boston Scientific Scimed, Inc. | Medical devices and methods of making the same |
| US8052745B2 (en) | 2007-09-13 | 2011-11-08 | Boston Scientific Scimed, Inc. | Endoprosthesis |
| US8052743B2 (en) | 2006-08-02 | 2011-11-08 | Boston Scientific Scimed, Inc. | Endoprosthesis with three-dimensional disintegration control |
| US8057534B2 (en) | 2006-09-15 | 2011-11-15 | Boston Scientific Scimed, Inc. | Bioerodible endoprostheses and methods of making the same |
| US8066763B2 (en) | 1998-04-11 | 2011-11-29 | Boston Scientific Scimed, Inc. | Drug-releasing stent with ceramic-containing layer |
| US8067054B2 (en) | 2007-04-05 | 2011-11-29 | Boston Scientific Scimed, Inc. | Stents with ceramic drug reservoir layer and methods of making and using the same |
| US8070797B2 (en) | 2007-03-01 | 2011-12-06 | Boston Scientific Scimed, Inc. | Medical device with a porous surface for delivery of a therapeutic agent |
| US8071156B2 (en) | 2009-03-04 | 2011-12-06 | Boston Scientific Scimed, Inc. | Endoprostheses |
| US8080055B2 (en) | 2006-12-28 | 2011-12-20 | Boston Scientific Scimed, Inc. | Bioerodible endoprostheses and methods of making the same |
| US8089029B2 (en) | 2006-02-01 | 2012-01-03 | Boston Scientific Scimed, Inc. | Bioabsorbable metal medical device and method of manufacture |
| US8128689B2 (en) | 2006-09-15 | 2012-03-06 | Boston Scientific Scimed, Inc. | Bioerodible endoprosthesis with biostable inorganic layers |
| US8187620B2 (en) | 2006-03-27 | 2012-05-29 | Boston Scientific Scimed, Inc. | Medical devices comprising a porous metal oxide or metal material and a polymer coating for delivering therapeutic agents |
| US8216632B2 (en) | 2007-11-02 | 2012-07-10 | Boston Scientific Scimed, Inc. | Endoprosthesis coating |
| US8221822B2 (en) | 2007-07-31 | 2012-07-17 | Boston Scientific Scimed, Inc. | Medical device coating by laser cladding |
| US8231980B2 (en) | 2008-12-03 | 2012-07-31 | Boston Scientific Scimed, Inc. | Medical implants including iridium oxide |
| US8236046B2 (en) | 2008-06-10 | 2012-08-07 | Boston Scientific Scimed, Inc. | Bioerodible endoprosthesis |
| US8267992B2 (en) | 2009-03-02 | 2012-09-18 | Boston Scientific Scimed, Inc. | Self-buffering medical implants |
| US8287937B2 (en) | 2009-04-24 | 2012-10-16 | Boston Scientific Scimed, Inc. | Endoprosthese |
| US8303643B2 (en) | 2001-06-27 | 2012-11-06 | Remon Medical Technologies Ltd. | Method and device for electrochemical formation of therapeutic species in vivo |
| US8313521B2 (en) | 1995-06-07 | 2012-11-20 | Cook Medical Technologies Llc | Method of delivering an implantable medical device with a bioabsorbable coating |
| US8353949B2 (en) | 2006-09-14 | 2013-01-15 | Boston Scientific Scimed, Inc. | Medical devices with drug-eluting coating |
| US8382824B2 (en) | 2008-10-03 | 2013-02-26 | Boston Scientific Scimed, Inc. | Medical implant having NANO-crystal grains with barrier layers of metal nitrides or fluorides |
| US8431149B2 (en) | 2007-03-01 | 2013-04-30 | Boston Scientific Scimed, Inc. | Coated medical devices for abluminal drug delivery |
| US8480620B2 (en) | 2009-12-11 | 2013-07-09 | Abbott Cardiovascular Systems Inc. | Coatings with tunable solubility profile for drug-coated balloon |
| US8501213B2 (en) | 2004-03-19 | 2013-08-06 | Abbott Laboratories | Multiple drug delivery from a balloon and a prosthesis |
| US8574615B2 (en) | 2006-03-24 | 2013-11-05 | Boston Scientific Scimed, Inc. | Medical devices having nanoporous coatings for controlled therapeutic agent delivery |
| US8642063B2 (en) | 2008-08-22 | 2014-02-04 | Cook Medical Technologies Llc | Implantable medical device coatings with biodegradable elastomer and releasable taxane agent |
| US8668732B2 (en) | 2010-03-23 | 2014-03-11 | Boston Scientific Scimed, Inc. | Surface treated bioerodible metal endoprostheses |
| US20140121750A1 (en) * | 2012-10-31 | 2014-05-01 | Cook Medical Technologies Llc | Fixation Process For Nesting Stents |
| US8771343B2 (en) | 2006-06-29 | 2014-07-08 | Boston Scientific Scimed, Inc. | Medical devices with selective titanium oxide coatings |
| US8808726B2 (en) | 2006-09-15 | 2014-08-19 | Boston Scientific Scimed. Inc. | Bioerodible endoprostheses and methods of making the same |
| US8815273B2 (en) | 2007-07-27 | 2014-08-26 | Boston Scientific Scimed, Inc. | Drug eluting medical devices having porous layers |
| US8815275B2 (en) | 2006-06-28 | 2014-08-26 | Boston Scientific Scimed, Inc. | Coatings for medical devices comprising a therapeutic agent and a metallic material |
| US8840660B2 (en) | 2006-01-05 | 2014-09-23 | Boston Scientific Scimed, Inc. | Bioerodible endoprostheses and methods of making the same |
| US8900292B2 (en) | 2007-08-03 | 2014-12-02 | Boston Scientific Scimed, Inc. | Coating for medical device having increased surface area |
| US8920491B2 (en) | 2008-04-22 | 2014-12-30 | Boston Scientific Scimed, Inc. | Medical devices having a coating of inorganic material |
| US8932346B2 (en) | 2008-04-24 | 2015-01-13 | Boston Scientific Scimed, Inc. | Medical devices having inorganic particle layers |
| US20160038317A1 (en) * | 2013-04-24 | 2016-02-11 | Vascular Dynamics, Inc. | Implantable vascular device having longitudinal struts |
| US9284409B2 (en) | 2007-07-19 | 2016-03-15 | Boston Scientific Scimed, Inc. | Endoprosthesis having a non-fouling surface |
| US9895158B2 (en) | 2007-10-26 | 2018-02-20 | University Of Virginia Patent Foundation | Method and apparatus for accelerated disintegration of blood clot |
| US20210308948A1 (en) * | 2012-01-24 | 2021-10-07 | Smith & Nephew, Inc. | Porous structure and methods of making same |
Families Citing this family (61)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7544192B2 (en) * | 2003-03-14 | 2009-06-09 | Sinexus, Inc. | Sinus delivery of sustained release therapeutics |
| WO2005087140A1 (en) | 2004-03-11 | 2005-09-22 | Percutaneous Cardiovascular Solutions Pty Limited | Percutaneous heart valve prosthesis |
| US20050251245A1 (en) * | 2004-05-05 | 2005-11-10 | Karl Sieradzki | Methods and apparatus with porous materials |
| KR100675379B1 (en) * | 2005-01-25 | 2007-01-29 | 삼성전자주식회사 | Printing system and printing method |
| RU2007140909A (en) * | 2005-04-04 | 2009-05-20 | Синексус, Инк. (Us) | DEVICE AND METHODS FOR TREATING DISEASES OF THE NANOLAIN SINUS |
| US8092520B2 (en) | 2005-11-10 | 2012-01-10 | CardiAQ Technologies, Inc. | Vascular prosthesis connecting stent |
| US8535707B2 (en) * | 2006-07-10 | 2013-09-17 | Intersect Ent, Inc. | Devices and methods for delivering active agents to the osteomeatal complex |
| WO2008013915A2 (en) * | 2006-07-28 | 2008-01-31 | Arshad Quadri | Percutaneous valve prosthesis and system and method for implanting same |
| US8221496B2 (en) | 2007-02-01 | 2012-07-17 | Cordis Corporation | Antithrombotic and anti-restenotic drug eluting stent |
| WO2009079418A2 (en) | 2007-12-18 | 2009-06-25 | Sinexus, Inc. | Self-expanding devices and methods therefor |
| CA2732355A1 (en) | 2008-08-01 | 2010-02-04 | Intersect Ent, Inc. | Methods and devices for crimping self-expanding devices |
| EP2367505B1 (en) | 2008-09-29 | 2020-08-12 | Edwards Lifesciences CardiAQ LLC | Heart valve |
| CA2739275C (en) | 2008-10-01 | 2017-01-17 | Impala, Inc. | Delivery system for vascular implant |
| CA2961053C (en) * | 2009-04-15 | 2019-04-30 | Edwards Lifesciences Cardiaq Llc | Vascular implant and delivery system |
| US10357640B2 (en) | 2009-05-15 | 2019-07-23 | Intersect Ent, Inc. | Expandable devices and methods for treating a nasal or sinus condition |
| US9283305B2 (en) | 2009-07-09 | 2016-03-15 | Medtronic Vascular, Inc. | Hollow tubular drug eluting medical devices |
| US20110070358A1 (en) | 2009-09-20 | 2011-03-24 | Medtronic Vascular, Inc. | Method of forming hollow tubular drug eluting medical devices |
| US8381774B2 (en) | 2009-09-20 | 2013-02-26 | Medtronic Vascular, Inc. | Methods for loading a drug eluting medical device |
| US8828474B2 (en) | 2009-09-20 | 2014-09-09 | Medtronic Vascular, Inc. | Apparatus and methods for loading a drug eluting medical device |
| US8678046B2 (en) | 2009-09-20 | 2014-03-25 | Medtronic Vascular, Inc. | Apparatus and methods for loading a drug eluting medical device |
| US20110313515A1 (en) | 2010-06-21 | 2011-12-22 | Arshad Quadri | Replacement heart valve |
| US9730790B2 (en) | 2009-09-29 | 2017-08-15 | Edwards Lifesciences Cardiaq Llc | Replacement valve and method |
| US8551159B2 (en) | 2010-04-01 | 2013-10-08 | Abbott Cardiovascular Systems Inc. | Implantable prosthesis having through-holes |
| US8562670B2 (en) | 2010-04-01 | 2013-10-22 | Abbott Cardiovascular Systems Inc. | Implantable prosthesis with depot retention feature |
| US8579964B2 (en) | 2010-05-05 | 2013-11-12 | Neovasc Inc. | Transcatheter mitral valve prosthesis |
| US8632846B2 (en) | 2010-09-17 | 2014-01-21 | Medtronic Vascular, Inc. | Apparatus and methods for loading a drug eluting medical device |
| US8333801B2 (en) | 2010-09-17 | 2012-12-18 | Medtronic Vascular, Inc. | Method of Forming a Drug-Eluting Medical Device |
| US8616040B2 (en) | 2010-09-17 | 2013-12-31 | Medtronic Vascular, Inc. | Method of forming a drug-eluting medical device |
| WO2012040655A2 (en) | 2010-09-23 | 2012-03-29 | Cardiaq Valve Technologies, Inc. | Replacement heart valves, delivery devices and methods |
| US20120239128A1 (en) * | 2011-03-16 | 2012-09-20 | Boston Scientific Scimed, Inc. | Stent and delivery system |
| USD665500S1 (en) | 2011-04-15 | 2012-08-14 | Novostent Corporation | Stent |
| US9308087B2 (en) | 2011-04-28 | 2016-04-12 | Neovasc Tiara Inc. | Sequentially deployed transcatheter mitral valve prosthesis |
| US9554897B2 (en) | 2011-04-28 | 2017-01-31 | Neovasc Tiara Inc. | Methods and apparatus for engaging a valve prosthesis with tissue |
| US9345573B2 (en) | 2012-05-30 | 2016-05-24 | Neovasc Tiara Inc. | Methods and apparatus for loading a prosthesis onto a delivery system |
| US10583002B2 (en) | 2013-03-11 | 2020-03-10 | Neovasc Tiara Inc. | Prosthetic valve with anti-pivoting mechanism |
| US20140277427A1 (en) | 2013-03-14 | 2014-09-18 | Cardiaq Valve Technologies, Inc. | Prosthesis for atraumatically grasping intralumenal tissue and methods of delivery |
| US9730791B2 (en) | 2013-03-14 | 2017-08-15 | Edwards Lifesciences Cardiaq Llc | Prosthesis for atraumatically grasping intralumenal tissue and methods of delivery |
| US10406332B2 (en) | 2013-03-14 | 2019-09-10 | Intersect Ent, Inc. | Systems, devices, and method for treating a sinus condition |
| US9681951B2 (en) | 2013-03-14 | 2017-06-20 | Edwards Lifesciences Cardiaq Llc | Prosthesis with outer skirt and anchors |
| US9486340B2 (en) | 2013-03-14 | 2016-11-08 | Medtronic Vascular, Inc. | Method for manufacturing a stent and stent manufactured thereby |
| US9572665B2 (en) | 2013-04-04 | 2017-02-21 | Neovasc Tiara Inc. | Methods and apparatus for delivering a prosthetic valve to a beating heart |
| DK2958607T3 (en) * | 2013-05-02 | 2016-08-29 | Cardionovum Gmbh | Ballonoverfladecoating |
| EP3016595B1 (en) | 2013-07-26 | 2018-12-19 | Edwards Lifesciences CardiAQ LLC | Systems for sealing openings in an anatomical wall |
| WO2015127283A1 (en) | 2014-02-21 | 2015-08-27 | Cardiaq Valve Technologies, Inc. | Delivery device for controlled deployement of a replacement valve |
| WO2015179423A1 (en) | 2014-05-19 | 2015-11-26 | Cardiaq Valve Technologies, Inc. | Replacement mitral valve with annular flap |
| US9532870B2 (en) | 2014-06-06 | 2017-01-03 | Edwards Lifesciences Corporation | Prosthetic valve for replacing a mitral valve |
| US10441416B2 (en) | 2015-04-21 | 2019-10-15 | Edwards Lifesciences Corporation | Percutaneous mitral valve replacement device |
| US10376363B2 (en) | 2015-04-30 | 2019-08-13 | Edwards Lifesciences Cardiaq Llc | Replacement mitral valve, delivery system for replacement mitral valve and methods of use |
| CA2990872C (en) | 2015-06-22 | 2022-03-22 | Edwards Lifescience Cardiaq Llc | Actively controllable heart valve implant and methods of controlling same |
| US10092400B2 (en) | 2015-06-23 | 2018-10-09 | Edwards Lifesciences Cardiaq Llc | Systems and methods for anchoring and sealing a prosthetic heart valve |
| US10575951B2 (en) | 2015-08-26 | 2020-03-03 | Edwards Lifesciences Cardiaq Llc | Delivery device and methods of use for transapical delivery of replacement mitral valve |
| US10117744B2 (en) | 2015-08-26 | 2018-11-06 | Edwards Lifesciences Cardiaq Llc | Replacement heart valves and methods of delivery |
| US10350066B2 (en) | 2015-08-28 | 2019-07-16 | Edwards Lifesciences Cardiaq Llc | Steerable delivery system for replacement mitral valve and methods of use |
| USD815744S1 (en) | 2016-04-28 | 2018-04-17 | Edwards Lifesciences Cardiaq Llc | Valve frame for a delivery system |
| US10350062B2 (en) | 2016-07-21 | 2019-07-16 | Edwards Lifesciences Corporation | Replacement heart valve prosthesis |
| WO2018035375A1 (en) | 2016-08-19 | 2018-02-22 | Edwards Lifesciences Corporation | Steerable delivery system for replacement mitral valve and methods of use |
| DK3503848T3 (en) | 2016-08-26 | 2021-11-15 | Edwards Lifesciences Corp | Replacement heart valve prosthesis in several parts |
| US10758348B2 (en) | 2016-11-02 | 2020-09-01 | Edwards Lifesciences Corporation | Supra and sub-annular mitral valve delivery system |
| US10813757B2 (en) | 2017-07-06 | 2020-10-27 | Edwards Lifesciences Corporation | Steerable rail delivery system |
| EP3720390A2 (en) | 2018-01-25 | 2020-10-14 | Edwards Lifesciences Corporation | Delivery system for aided replacement valve recapture and repositioning post- deployment |
| US11051934B2 (en) | 2018-02-28 | 2021-07-06 | Edwards Lifesciences Corporation | Prosthetic mitral valve with improved anchors and seal |
Citations (41)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5609629A (en) * | 1995-06-07 | 1997-03-11 | Med Institute, Inc. | Coated implantable medical device |
| US5797898A (en) * | 1996-07-02 | 1998-08-25 | Massachusetts Institute Of Technology | Microchip drug delivery devices |
| US5824049A (en) * | 1995-06-07 | 1998-10-20 | Med Institute, Inc. | Coated implantable medical device |
| US5972027A (en) * | 1997-09-30 | 1999-10-26 | Scimed Life Systems, Inc | Porous stent drug delivery system |
| US5992769A (en) * | 1995-06-09 | 1999-11-30 | The Regents Of The University Of Michigan | Microchannel system for fluid delivery |
| US6071305A (en) * | 1996-11-25 | 2000-06-06 | Alza Corporation | Directional drug delivery stent and method of use |
| US6156062A (en) * | 1997-12-03 | 2000-12-05 | Ave Connaught | Helically wrapped interlocking stent |
| US6206915B1 (en) * | 1998-09-29 | 2001-03-27 | Medtronic Ave, Inc. | Drug storing and metering stent |
| US6241762B1 (en) * | 1998-03-30 | 2001-06-05 | Conor Medsystems, Inc. | Expandable medical device with ductile hinges |
| US6254632B1 (en) * | 2000-09-28 | 2001-07-03 | Advanced Cardiovascular Systems, Inc. | Implantable medical device having protruding surface structures for drug delivery and cover attachment |
| US6273908B1 (en) * | 1997-10-24 | 2001-08-14 | Robert Ndondo-Lay | Stents |
| US6273913B1 (en) * | 1997-04-18 | 2001-08-14 | Cordis Corporation | Modified stent useful for delivery of drugs along stent strut |
| US6293967B1 (en) * | 1998-10-29 | 2001-09-25 | Conor Medsystems, Inc. | Expandable medical device with ductile hinges |
| US6299604B1 (en) * | 1998-08-20 | 2001-10-09 | Cook Incorporated | Coated implantable medical device |
| US20010029351A1 (en) * | 1998-04-16 | 2001-10-11 | Robert Falotico | Drug combinations and delivery devices for the prevention and treatment of vascular disease |
| US20020007209A1 (en) * | 2000-03-06 | 2002-01-17 | Scheerder Ivan De | Intraluminar perforated radially expandable drug delivery prosthesis and a method for the production thereof |
| US20020028243A1 (en) * | 1998-09-25 | 2002-03-07 | Masters David B. | Protein matrix materials, devices and methods of making and using thereof |
| US20020038145A1 (en) * | 2000-06-05 | 2002-03-28 | Jang G. David | Intravascular stent with increasing coating retaining capacity |
| US6379381B1 (en) * | 1999-09-03 | 2002-04-30 | Advanced Cardiovascular Systems, Inc. | Porous prosthesis and a method of depositing substances into the pores |
| US20020068969A1 (en) * | 2000-10-16 | 2002-06-06 | Shanley John F. | Expandable medical device with improved spatial distribution |
| US20020082680A1 (en) * | 2000-10-16 | 2002-06-27 | Shanley John F. | Expandable medical device for delivery of beneficial agent |
| US6423092B2 (en) * | 1999-12-22 | 2002-07-23 | Ethicon, Inc. | Biodegradable stent |
| US20020155212A1 (en) * | 2001-04-24 | 2002-10-24 | Hossainy Syed Faiyaz Ahmed | Coating for a stent and a method of forming the same |
| US6491666B1 (en) * | 1999-11-17 | 2002-12-10 | Microchips, Inc. | Microfabricated devices for the delivery of molecules into a carrier fluid |
| US6506437B1 (en) * | 2000-10-17 | 2003-01-14 | Advanced Cardiovascular Systems, Inc. | Methods of coating an implantable device having depots formed in a surface thereof |
| US20030028244A1 (en) * | 1995-06-07 | 2003-02-06 | Cook Incorporated | Coated implantable medical device |
| US20030036794A1 (en) * | 1995-06-07 | 2003-02-20 | Cook Incorporated | Coated implantable medical device |
| US20030060877A1 (en) * | 2001-09-25 | 2003-03-27 | Robert Falotico | Coated medical devices for the treatment of vascular disease |
| US20030068355A1 (en) * | 2001-08-20 | 2003-04-10 | Shanley John F. | Therapeutic agent delivery device with protective separating layer |
| US6551838B2 (en) * | 2000-03-02 | 2003-04-22 | Microchips, Inc. | Microfabricated devices for the storage and selective exposure of chemicals and devices |
| US6558733B1 (en) * | 2000-10-26 | 2003-05-06 | Advanced Cardiovascular Systems, Inc. | Method for etching a micropatterned microdepot prosthesis |
| US6635082B1 (en) * | 2000-12-29 | 2003-10-21 | Advanced Cardiovascular Systems Inc. | Radiopaque stent |
| US20030199970A1 (en) * | 1998-03-30 | 2003-10-23 | Conor Medsystems, Inc. | Expandable medical device for delivery of beneficial agent |
| US20030216699A1 (en) * | 2000-05-12 | 2003-11-20 | Robert Falotico | Coated medical devices for the prevention and treatment of vascular disease |
| US6699281B2 (en) * | 2001-07-20 | 2004-03-02 | Sorin Biomedica Cardio S.P.A. | Angioplasty stents |
| US6730116B1 (en) * | 1999-04-16 | 2004-05-04 | Medtronic, Inc. | Medical device for intraluminal endovascular stenting |
| US6752829B2 (en) * | 2001-01-30 | 2004-06-22 | Scimed Life Systems, Inc. | Stent with channel(s) for containing and delivering a biologically active material and method for manufacturing the same |
| US20040127976A1 (en) * | 2002-09-20 | 2004-07-01 | Conor Medsystems, Inc. | Method and apparatus for loading a beneficial agent into an expandable medical device |
| US20040127977A1 (en) * | 2002-09-20 | 2004-07-01 | Conor Medsystems, Inc. | Expandable medical device with openings for delivery of multiple beneficial agents |
| US6758859B1 (en) * | 2000-10-30 | 2004-07-06 | Kenny L. Dang | Increased drug-loading and reduced stress drug delivery device |
| US20040144506A1 (en) * | 2002-10-17 | 2004-07-29 | Bos Gmbh & Co. Kg | Window shade with extraction slot cover |
Family Cites Families (235)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US516608A (en) * | 1894-03-13 | Window-screen | ||
| US3657744A (en) | 1970-05-08 | 1972-04-25 | Univ Minnesota | Method for fixing prosthetic implants in a living body |
| US5876419A (en) | 1976-10-02 | 1999-03-02 | Navius Corporation | Stent and method for making a stent |
| US5643314A (en) | 1995-11-13 | 1997-07-01 | Navius Corporation | Self-expanding stent |
| US4300244A (en) | 1979-09-19 | 1981-11-17 | Carbomedics, Inc. | Cardiovascular grafts |
| US4531936A (en) | 1981-01-29 | 1985-07-30 | Gordon Robert T | Device and method for the selective delivery of drugs to the myocardium |
| US5441745A (en) | 1982-03-30 | 1995-08-15 | Vestar, Inc. | Method of delivering micellular particles encapsulating chemotherapeutic agents to tumors in a body |
| US4542025A (en) | 1982-07-29 | 1985-09-17 | The Stolle Research And Development Corporation | Injectable, long-acting microparticle formulation for the delivery of anti-inflammatory agents |
| US4834755A (en) | 1983-04-04 | 1989-05-30 | Pfizer Hospital Products Group, Inc. | Triaxially-braided fabric prosthesis |
| US4580568A (en) | 1984-10-01 | 1986-04-08 | Cook, Incorporated | Percutaneous endovascular stent and method for insertion thereof |
| US4824436A (en) | 1985-04-09 | 1989-04-25 | Harvey Wolinsky | Method for the prevention of restenosis |
| US4650466A (en) | 1985-11-01 | 1987-03-17 | Angiobrade Partners | Angioplasty device |
| US5102417A (en) | 1985-11-07 | 1992-04-07 | Expandable Grafts Partnership | Expandable intraluminal graft, and method and apparatus for implanting an expandable intraluminal graft |
| US4733665C2 (en) | 1985-11-07 | 2002-01-29 | Expandable Grafts Partnership | Expandable intraluminal graft and method and apparatus for implanting an expandable intraluminal graft |
| US4955878A (en) | 1986-04-04 | 1990-09-11 | Biotechnology, Inc. | Kit for preventing or treating arterial dysfunction resulting from angioplasty procedures |
| JPH0763489B2 (en) | 1986-10-31 | 1995-07-12 | 宇部興産株式会社 | Medical tube |
| US4800882A (en) | 1987-03-13 | 1989-01-31 | Cook Incorporated | Endovascular stent and delivery system |
| US5059211A (en) | 1987-06-25 | 1991-10-22 | Duke University | Absorbable vascular stent |
| US4969458A (en) | 1987-07-06 | 1990-11-13 | Medtronic, Inc. | Intracoronary stent and method of simultaneous angioplasty and stent implant |
| US5460817A (en) | 1988-01-19 | 1995-10-24 | Allied Colloids Ltd. | Particulate composition comprising a core of matrix polymer with active ingredient distributed therein |
| US4989601A (en) | 1988-05-02 | 1991-02-05 | Medical Engineering & Development Institute, Inc. | Method, apparatus, and substance for treating tissue having neoplastic cells |
| JPH0255064A (en) | 1988-08-03 | 1990-02-23 | Toa O | Skin removal for throm bus in blood vessel using catheter and throm bus removing system in blood vessel using catheter |
| US5213580A (en) | 1988-08-24 | 1993-05-25 | Endoluminal Therapeutics, Inc. | Biodegradable polymeric endoluminal sealing process |
| DE68922497T2 (en) | 1988-08-24 | 1995-09-14 | Marvin J Slepian | ENDOLUMINAL SEAL WITH BISDEGRADABLE POLYMERS. |
| US5019090A (en) | 1988-09-01 | 1991-05-28 | Corvita Corporation | Radially expandable endoprosthesis and the like |
| US5053048A (en) | 1988-09-22 | 1991-10-01 | Cordis Corporation | Thromboresistant coating |
| CA1322628C (en) | 1988-10-04 | 1993-10-05 | Richard A. Schatz | Expandable intraluminal graft |
| LU87410A1 (en) | 1988-12-20 | 1990-07-10 | Cird | COSMETIC OR PHARMACEUTICAL COMPOSITION CONTAINING POLYMERIC OR FATTY BODY MICROSPHERES CHARGED WITH AT LEAST ONE ACTIVE PRODUCT |
| CH678393A5 (en) | 1989-01-26 | 1991-09-13 | Ulrich Prof Dr Med Sigwart | |
| US5078726A (en) | 1989-02-01 | 1992-01-07 | Kreamer Jeffry W | Graft stent and method of repairing blood vessels |
| US4960790A (en) | 1989-03-09 | 1990-10-02 | University Of Kansas | Derivatives of taxol, pharmaceutical compositions thereof and methods for the preparation thereof |
| WO1990013332A1 (en) | 1989-05-11 | 1990-11-15 | Cedars-Sinai Medical Center | Stent with sustained drug delivery |
| US4990155A (en) | 1989-05-19 | 1991-02-05 | Wilkoff Howard M | Surgical stent method and apparatus |
| US4994071A (en) | 1989-05-22 | 1991-02-19 | Cordis Corporation | Bifurcating stent apparatus and method |
| US5171262A (en) | 1989-06-15 | 1992-12-15 | Cordis Corporation | Non-woven endoprosthesis |
| US5674278A (en) | 1989-08-24 | 1997-10-07 | Arterial Vascular Engineering, Inc. | Endovascular support device |
| US5176617A (en) | 1989-12-11 | 1993-01-05 | Medical Innovative Technologies R & D Limited Partnership | Use of a stent with the capability to inhibit malignant growth in a vessel such as a biliary duct |
| US5059166A (en) | 1989-12-11 | 1991-10-22 | Medical Innovative Technologies R & D Limited Partnership | Intra-arterial stent with the capability to inhibit intimal hyperplasia |
| US5439446A (en) | 1994-06-30 | 1995-08-08 | Boston Scientific Corporation | Stent and therapeutic delivery system |
| US5304121A (en) | 1990-12-28 | 1994-04-19 | Boston Scientific Corporation | Drug delivery system making use of a hydrogel polymer coating |
| US5049132A (en) | 1990-01-08 | 1991-09-17 | Cordis Corporation | Balloon catheter for delivering therapeutic agents |
| US5192744A (en) | 1990-01-12 | 1993-03-09 | Northwestern University | Method of inhibiting angiogenesis of tumors |
| DK0512071T3 (en) | 1990-01-25 | 1996-11-25 | Childrens Hospital | Methods and compositions for inhibiting angiogenesis |
| DK0441516T3 (en) | 1990-02-08 | 1995-06-12 | Howmedica | Inflatable catheter |
| DE69110787T2 (en) | 1990-02-28 | 1996-04-04 | Medtronic Inc | INTRALUMINAL PROSTHESIS WITH ACTIVE ELEMENTATION. |
| US5545208A (en) | 1990-02-28 | 1996-08-13 | Medtronic, Inc. | Intralumenal drug eluting prosthesis |
| US5242399A (en) | 1990-04-25 | 1993-09-07 | Advanced Cardiovascular Systems, Inc. | Method and system for stent delivery |
| US5344426A (en) | 1990-04-25 | 1994-09-06 | Advanced Cardiovascular Systems, Inc. | Method and system for stent delivery |
| US5092841A (en) | 1990-05-17 | 1992-03-03 | Wayne State University | Method for treating an arterial wall injured during angioplasty |
| AU7998091A (en) | 1990-05-17 | 1991-12-10 | Harbor Medical Devices, Inc. | Medical device polymer |
| EP0528993A1 (en) | 1990-05-18 | 1993-03-03 | STACK, Richard S. | Intraluminal stent |
| US5407683A (en) | 1990-06-01 | 1995-04-18 | Research Corporation Technologies, Inc. | Pharmaceutical solutions and emulsions containing taxol |
| EP0533816B1 (en) | 1990-06-15 | 1995-06-14 | Cortrak Medical, Inc. | Drug delivery apparatus |
| DE69129812T2 (en) | 1990-07-12 | 1999-02-11 | Sts Biopolymers Inc | ANTITHROMOGIC AND / OR ANTIMICROBIAL COMPOSITION |
| EP0470569B1 (en) | 1990-08-08 | 1995-11-22 | Takeda Chemical Industries, Ltd. | Intravascular embolizing agent containing angiogenesis inhibiting substance |
| DE4035809A1 (en) | 1990-11-10 | 1992-05-14 | Boehringer Mannheim Gmbh | USE OF THIAZOLOISOINDOLINONE DERIVATIVES AS ANTIVIRAL MEDICAMENTS |
| AU1579092A (en) | 1991-02-27 | 1992-10-06 | Nova Pharmaceutical Corporation | Anti-infective and anti-inflammatory releasing systems for medical devices |
| US5171217A (en) | 1991-02-28 | 1992-12-15 | Indiana University Foundation | Method for delivery of smooth muscle cell inhibitors |
| US5197978B1 (en) | 1991-04-26 | 1996-05-28 | Advanced Coronary Tech | Removable heat-recoverable tissue supporting device |
| FR2678833B1 (en) | 1991-07-08 | 1995-04-07 | Rhone Poulenc Rorer Sa | NEW PHARMACEUTICAL COMPOSITIONS BASED ON DERIVATIVES OF THE TAXANE CLASS. |
| US5811447A (en) | 1993-01-28 | 1998-09-22 | Neorx Corporation | Therapeutic inhibitor of vascular smooth muscle cells |
| US6515009B1 (en) | 1991-09-27 | 2003-02-04 | Neorx Corporation | Therapeutic inhibitor of vascular smooth muscle cells |
| US5500013A (en) | 1991-10-04 | 1996-03-19 | Scimed Life Systems, Inc. | Biodegradable drug delivery vascular stent |
| US5464450A (en) | 1991-10-04 | 1995-11-07 | Scimed Lifesystems Inc. | Biodegradable drug delivery vascular stent |
| WO1993006792A1 (en) | 1991-10-04 | 1993-04-15 | Scimed Life Systems, Inc. | Biodegradable drug delivery vascular stent |
| CA2079417C (en) | 1991-10-28 | 2003-01-07 | Lilip Lau | Expandable stents and method of making same |
| FR2683449A1 (en) | 1991-11-08 | 1993-05-14 | Cardon Alain | ENDOPROTHESIS FOR TRANSLUMINAL IMPLANTATION. |
| US5270047A (en) | 1991-11-21 | 1993-12-14 | Kauffman Raymond F | Local delivery of dipyridamole for the treatment of proliferative diseases |
| EP0643706A1 (en) | 1991-11-27 | 1995-03-22 | Zynaxis Inc. | Compounds, compositions and methods for binding bio-affecting substances to surface membranes of bio-particles |
| GB2262365B (en) | 1991-12-10 | 1995-08-09 | Sony Broadcast & Communication | Apparatus and methods for designing,analyzing or simulating signal processing functions |
| US5260002A (en) | 1991-12-23 | 1993-11-09 | Vanderbilt University | Method and apparatus for producing uniform polymeric spheres |
| CA2087132A1 (en) | 1992-01-31 | 1993-08-01 | Michael S. Williams | Stent capable of attachment within a body lumen |
| US5282823A (en) | 1992-03-19 | 1994-02-01 | Medtronic, Inc. | Intravascular radially expandable stent |
| DE4214215A1 (en) | 1992-04-30 | 1993-11-04 | Behringwerke Ag | USE OF INHIBITORS OF PLASMINOGEN ACTIVATORS FOR TREATING INFLAMMATION |
| US5383928A (en) | 1992-06-10 | 1995-01-24 | Emory University | Stent sheath for local drug delivery |
| GB9213077D0 (en) | 1992-06-19 | 1992-08-05 | Erba Carlo Spa | Polymerbound taxol derivatives |
| KR940003548U (en) | 1992-08-14 | 1994-02-21 | 김형술 | Laundry dryer |
| US5650447A (en) | 1992-08-24 | 1997-07-22 | The United States Of America As Represented By The Secretary Of The Department Of Health And Human Services | Nitric oxide-releasing polymers to treat restenosis and related disorders |
| US5342621A (en) | 1992-09-15 | 1994-08-30 | Advanced Cardiovascular Systems, Inc. | Antithrombogenic surface |
| US5770609A (en) | 1993-01-28 | 1998-06-23 | Neorx Corporation | Prevention and treatment of cardiovascular pathologies |
| EP0752885B1 (en) | 1992-09-25 | 2003-07-09 | Neorx Corporation | Therapeutic inhibitor of vascular smooth muscle cells |
| US5578075B1 (en) | 1992-11-04 | 2000-02-08 | Daynke Res Inc | Minimally invasive bioactivated endoprosthesis for vessel repair |
| US5419760A (en) | 1993-01-08 | 1995-05-30 | Pdt Systems, Inc. | Medicament dispensing stent for prevention of restenosis of a blood vessel |
| ES2166370T3 (en) | 1993-01-19 | 2002-04-16 | Schneider Usa Inc | IMPLANTABLE FILAMENT IN COMPOSITE MATERIAL. |
| US5981568A (en) | 1993-01-28 | 1999-11-09 | Neorx Corporation | Therapeutic inhibitor of vascular smooth muscle cells |
| US6663881B2 (en) | 1993-01-28 | 2003-12-16 | Neorx Corporation | Therapeutic inhibitor of vascular smooth muscle cells |
| US5595722A (en) | 1993-01-28 | 1997-01-21 | Neorx Corporation | Method for identifying an agent which increases TGF-beta levels |
| US6491938B2 (en) | 1993-05-13 | 2002-12-10 | Neorx Corporation | Therapeutic inhibitor of vascular smooth muscle cells |
| US5439686A (en) | 1993-02-22 | 1995-08-08 | Vivorx Pharmaceuticals, Inc. | Methods for in vivo delivery of substantially water insoluble pharmacologically active agents and compositions useful therefor |
| WO1994021308A1 (en) | 1993-03-18 | 1994-09-29 | Cedars-Sinai Medical Center | Drug incorporating and releasing polymeric coating for bioprosthesis |
| WO1994024961A1 (en) | 1993-04-23 | 1994-11-10 | Schneider (Usa) Inc. | Covered stent and stent delivery device |
| US5441515A (en) | 1993-04-23 | 1995-08-15 | Advanced Cardiovascular Systems, Inc. | Ratcheting stent |
| ATE164056T1 (en) | 1993-04-23 | 1998-04-15 | Schneider Europ Ag | STENT HAVING A COATING OF ELASTIC MATERIAL AND METHOD FOR APPLYING THE COATING TO THE STENT |
| US5464650A (en) | 1993-04-26 | 1995-11-07 | Medtronic, Inc. | Intravascular stent and method |
| US5849035A (en) | 1993-04-28 | 1998-12-15 | Focal, Inc. | Methods for intraluminal photothermoforming |
| CA2162586C (en) | 1993-05-13 | 2006-01-03 | David J. Grainger | Prevention and treatment of pathologies associated with abnormally proliferative smooth muscle cells |
| US5994341A (en) | 1993-07-19 | 1999-11-30 | Angiogenesis Technologies, Inc. | Anti-angiogenic Compositions and methods for the treatment of arthritis |
| CN100998869A (en) | 1993-07-19 | 2007-07-18 | 血管技术药物公司 | Anti-angiogene compositions and methods of use |
| DE4325435A1 (en) | 1993-07-29 | 1995-02-02 | Basf Ag | New combination of active ingredients |
| EP0711158B2 (en) | 1993-07-29 | 2008-07-23 | THE GOVERNMENT OF THE UNITED STATES OF AMERICA, as represented by THE SECRETARY, DEPARTMENT OF HEALTH AND HUMAN SERVICES | Method of treating atherosclerosis or restenosis using microtubule stabilizing agent |
| US5380299A (en) | 1993-08-30 | 1995-01-10 | Med Institute, Inc. | Thrombolytic treated intravascular medical device |
| US5457113A (en) | 1993-10-15 | 1995-10-10 | Eli Lilly And Company | Methods for inhibiting vascular smooth muscle cell proliferation and restinosis |
| US5415869A (en) | 1993-11-12 | 1995-05-16 | The Research Foundation Of State University Of New York | Taxol formulation |
| US5443497A (en) | 1993-11-22 | 1995-08-22 | The Johns Hopkins University | Percutaneous prosthetic by-pass graft and method of use |
| JP3033412B2 (en) * | 1993-11-26 | 2000-04-17 | 株式会社デンソー | Method for manufacturing semiconductor device |
| US5556413A (en) | 1994-03-11 | 1996-09-17 | Advanced Cardiovascular Systems, Inc. | Coiled stent with locking ends |
| US5843120A (en) | 1994-03-17 | 1998-12-01 | Medinol Ltd. | Flexible-expandable stent |
| US5449373A (en) | 1994-03-17 | 1995-09-12 | Medinol Ltd. | Articulated stent |
| US5733303A (en) | 1994-03-17 | 1998-03-31 | Medinol Ltd. | Flexible expandable stent |
| EP0766539B1 (en) | 1994-06-13 | 2003-05-21 | Endomed, Inc. | Expandable endovascular graft and method for forming the same |
| US5545210A (en) | 1994-09-22 | 1996-08-13 | Advanced Coronary Technology, Inc. | Method of implanting a permanent shape memory alloy stent |
| US5817152A (en) | 1994-10-19 | 1998-10-06 | Birdsall; Matthew | Connected stent apparatus |
| AU3783295A (en) | 1994-11-16 | 1996-05-23 | Advanced Cardiovascular Systems Inc. | Shape memory locking mechanism for intravascular stent |
| CA2163824C (en) | 1994-11-28 | 2000-06-20 | Richard J. Saunders | Method and apparatus for direct laser cutting of metal stents |
| US5665591A (en) | 1994-12-06 | 1997-09-09 | Trustees Of Boston University | Regulation of smooth muscle cell proliferation |
| GB9505721D0 (en) | 1995-03-21 | 1995-05-10 | Univ London | Expandable surgical stent |
| US5605696A (en) | 1995-03-30 | 1997-02-25 | Advanced Cardiovascular Systems, Inc. | Drug loaded polymeric material and method of manufacture |
| EP0734698B9 (en) | 1995-04-01 | 2006-07-05 | Variomed AG | Stent for transluminal implantation into hollow organs |
| US5837313A (en) | 1995-04-19 | 1998-11-17 | Schneider (Usa) Inc | Drug release stent coating process |
| US7550005B2 (en) | 1995-06-07 | 2009-06-23 | Cook Incorporated | Coated implantable medical device |
| HU221910B1 (en) | 1995-07-25 | 2003-02-28 | Medstent Inc. | Expandible stent |
| DE19539449A1 (en) | 1995-10-24 | 1997-04-30 | Biotronik Mess & Therapieg | Process for the production of intraluminal stents from bioresorbable polymer material |
| US5607442A (en) | 1995-11-13 | 1997-03-04 | Isostent, Inc. | Stent with improved radiopacity and appearance characteristics |
| US5741293A (en) | 1995-11-28 | 1998-04-21 | Wijay; Bandula | Locking stent |
| US6203569B1 (en) | 1996-01-04 | 2001-03-20 | Bandula Wijay | Flexible stent |
| US6017363A (en) | 1997-09-22 | 2000-01-25 | Cordis Corporation | Bifurcated axially flexible stent |
| US5843117A (en) | 1996-02-14 | 1998-12-01 | Inflow Dynamics Inc. | Implantable vascular and endoluminal stents and process of fabricating the same |
| US6441025B2 (en) | 1996-03-12 | 2002-08-27 | Pg-Txl Company, L.P. | Water soluble paclitaxel derivatives |
| US5725548A (en) | 1996-04-08 | 1998-03-10 | Iowa India Investments Company Limited | Self-locking stent and method for its production |
| US5713949A (en) | 1996-08-06 | 1998-02-03 | Jayaraman; Swaminathan | Microporous covered stents and method of coating |
| US5928916A (en) | 1996-04-25 | 1999-07-27 | Medtronic, Inc. | Ionic attachment of biomolecules with a guanidino moiety to medical device surfaces |
| EP0927006B1 (en) | 1996-04-26 | 2006-01-18 | Boston Scientific Scimed, Inc. | Intravascular stent |
| US5922021A (en) | 1996-04-26 | 1999-07-13 | Jang; G. David | Intravascular stent |
| US5617878A (en) | 1996-05-31 | 1997-04-08 | Taheri; Syde A. | Stent and method for treatment of aortic occlusive disease |
| US5697971A (en) | 1996-06-11 | 1997-12-16 | Fischell; Robert E. | Multi-cell stent with cells having differing characteristics |
| US5922020A (en) | 1996-08-02 | 1999-07-13 | Localmed, Inc. | Tubular prosthesis having improved expansion and imaging characteristics |
| US6088192A (en) | 1996-08-05 | 2000-07-11 | Quantum Corporation | Roll-biased head suspension for reduced track misregistration |
| US6007517A (en) | 1996-08-19 | 1999-12-28 | Anderson; R. David | Rapid exchange/perfusion angioplasty catheter |
| US5776183A (en) | 1996-08-23 | 1998-07-07 | Kanesaka; Nozomu | Expandable stent |
| US6057367A (en) | 1996-08-30 | 2000-05-02 | Duke University | Manipulating nitrosative stress to kill pathologic microbes, pathologic helminths and pathologically proliferating cells or to upregulate nitrosative stress defenses |
| US5807404A (en) | 1996-09-19 | 1998-09-15 | Medinol Ltd. | Stent with variable features to optimize support and method of making such stent |
| US5824045A (en) | 1996-10-21 | 1998-10-20 | Inflow Dynamics Inc. | Vascular and endoluminal stents |
| US5868781A (en) | 1996-10-22 | 1999-02-09 | Scimed Life Systems, Inc. | Locking stent |
| AU4896797A (en) | 1996-11-04 | 1998-05-29 | Davidson, Charles | Extendible stent apparatus and method for deploying the same |
| US5980972A (en) | 1996-12-20 | 1999-11-09 | Schneider (Usa) Inc | Method of applying drug-release coatings |
| IT1289815B1 (en) | 1996-12-30 | 1998-10-16 | Sorin Biomedica Cardio Spa | ANGIOPLASTIC STENT AND RELATED PRODUCTION PROCESS |
| US5733330A (en) | 1997-01-13 | 1998-03-31 | Advanced Cardiovascular Systems, Inc. | Balloon-expandable, crush-resistant locking stent |
| US5980551A (en) | 1997-02-07 | 1999-11-09 | Endovasc Ltd., Inc. | Composition and method for making a biodegradable drug delivery stent |
| KR100526913B1 (en) | 1997-02-20 | 2005-11-09 | 쿡 인코포레이티드 | Coated implantable medical device |
| US5853419A (en) | 1997-03-17 | 1998-12-29 | Surface Genesis, Inc. | Stent |
| US5843172A (en) | 1997-04-15 | 1998-12-01 | Advanced Cardiovascular Systems, Inc. | Porous medicated stent |
| US6240616B1 (en) | 1997-04-15 | 2001-06-05 | Advanced Cardiovascular Systems, Inc. | Method of manufacturing a medicated porous metal prosthesis |
| US5843175A (en) | 1997-06-13 | 1998-12-01 | Global Therapeutics, Inc. | Enhanced flexibility surgical stent |
| FR2764794B1 (en) | 1997-06-20 | 1999-11-12 | Nycomed Lab Sa | EXPANDED TUBULAR DEVICE WITH VARIABLE THICKNESS |
| CA2241558A1 (en) | 1997-06-24 | 1998-12-24 | Advanced Cardiovascular Systems, Inc. | Stent with reinforced struts and bimodal deployment |
| US5855600A (en) | 1997-08-01 | 1999-01-05 | Inflow Dynamics Inc. | Flexible implantable stent with composite design |
| US5899935A (en) | 1997-08-04 | 1999-05-04 | Schneider (Usa) Inc. | Balloon expandable braided stent with restraint |
| US5984957A (en) | 1997-08-12 | 1999-11-16 | Schneider (Usa) Inc | Radially expanded prostheses with axial diameter control |
| US6306166B1 (en) | 1997-08-13 | 2001-10-23 | Scimed Life Systems, Inc. | Loading and release of water-insoluble drugs |
| US6165195A (en) | 1997-08-13 | 2000-12-26 | Advanced Cardiovascylar Systems, Inc. | Stent and catheter assembly and method for treating bifurcations |
| WO1999009912A1 (en) | 1997-08-26 | 1999-03-04 | Technion Research And Development Foundation Ltd. | Intravascular apparatus and method |
| DE69838256T2 (en) | 1997-09-24 | 2008-05-15 | Med Institute, Inc., West Lafayette | RADIAL EXPANDABLE STENT |
| DE19743373A1 (en) | 1997-09-30 | 1999-04-15 | Univ Heidelberg | · 3 ·· 2 · P-polyphosphazene |
| US5976182A (en) | 1997-10-03 | 1999-11-02 | Advanced Cardiovascular Systems, Inc. | Balloon-expandable, crush-resistant locking stent and method of loading the same |
| US6309414B1 (en) | 1997-11-04 | 2001-10-30 | Sorin Biomedica Cardio S.P.A. | Angioplasty stents |
| US6030414A (en) | 1997-11-13 | 2000-02-29 | Taheri; Syde A. | Variable stent and method for treatment of arterial disease |
| US5964798A (en) | 1997-12-16 | 1999-10-12 | Cardiovasc, Inc. | Stent having high radial strength |
| US6077296A (en) | 1998-03-04 | 2000-06-20 | Endologix, Inc. | Endoluminal vascular prosthesis |
| US6132461A (en) | 1998-03-27 | 2000-10-17 | Intratherapeutics, Inc. | Stent with dual support structure |
| EP1222941B2 (en) | 1998-03-30 | 2009-04-22 | Conor Medsystems, Inc. | Flexible medical device |
| US6019789A (en) | 1998-04-01 | 2000-02-01 | Quanam Medical Corporation | Expandable unit cell and intraluminal stent |
| WO1999055396A1 (en) | 1998-04-27 | 1999-11-04 | Surmodics, Inc. | Bioactive agent release coating |
| AU5947299A (en) | 1998-04-28 | 1999-11-16 | American National Red Cross, The | Method of determining osteogenic potential of human demineralized bone matrix powder |
| US6013099A (en) | 1998-04-29 | 2000-01-11 | Medtronic, Inc. | Medical device for delivering a water-insoluble therapeutic salt or substance |
| US6280411B1 (en) | 1998-05-18 | 2001-08-28 | Scimed Life Systems, Inc. | Localized delivery of drug agents |
| US6083258A (en) | 1998-05-28 | 2000-07-04 | Yadav; Jay S. | Locking stent |
| AU5054499A (en) | 1998-07-21 | 2000-02-14 | Biocompatibles Limited | Coating |
| US6203991B1 (en) | 1998-08-21 | 2001-03-20 | The Regents Of The University Of Michigan | Inhibition of smooth muscle cell migration by heme oxygenase I |
| US6245104B1 (en) * | 1999-02-28 | 2001-06-12 | Inflow Dynamics Inc. | Method of fabricating a biocompatible stent |
| EP1140243B1 (en) | 1998-12-31 | 2003-04-09 | Angiotech Pharmaceuticals, Inc. | Stent grafts with bioactive coatings |
| US6419692B1 (en) * | 1999-02-03 | 2002-07-16 | Scimed Life Systems, Inc. | Surface protection method for stents and balloon catheters for drug delivery |
| US6245101B1 (en) | 1999-05-03 | 2001-06-12 | William J. Drasler | Intravascular hinge stent |
| US6375676B1 (en) | 1999-05-17 | 2002-04-23 | Advanced Cardiovascular Systems, Inc. | Self-expanding stent with enhanced delivery precision and stent delivery system |
| US6290673B1 (en) | 1999-05-20 | 2001-09-18 | Conor Medsystems, Inc. | Expandable medical device delivery system and method |
| US6368346B1 (en) | 1999-06-03 | 2002-04-09 | American Medical Systems, Inc. | Bioresorbable stent |
| US6713119B2 (en) | 1999-09-03 | 2004-03-30 | Advanced Cardiovascular Systems, Inc. | Biocompatible coating for a prosthesis and a method of forming the same |
| EP1214108B1 (en) | 1999-09-03 | 2007-01-10 | Advanced Cardiovascular Systems, Inc. | A porous prosthesis and a method of depositing substances into the pores |
| US6759054B2 (en) | 1999-09-03 | 2004-07-06 | Advanced Cardiovascular Systems, Inc. | Ethylene vinyl alcohol composition and coating |
| US6239118B1 (en) | 1999-10-05 | 2001-05-29 | Richard A. Schatz | Method for preventing restenosis using a substituted adenine derivative |
| US6682545B1 (en) | 1999-10-06 | 2004-01-27 | The Penn State Research Foundation | System and device for preventing restenosis in body vessels |
| WO2001026584A1 (en) | 1999-10-14 | 2001-04-19 | United Stenting, Inc. | Stents with multilayered struts |
| US6613432B2 (en) | 1999-12-22 | 2003-09-02 | Biosurface Engineering Technologies, Inc. | Plasma-deposited coatings, devices and methods |
| US6471979B2 (en) | 1999-12-29 | 2002-10-29 | Estrogen Vascular Technology, Llc | Apparatus and method for delivering compounds to a living organism |
| US6899731B2 (en) | 1999-12-30 | 2005-05-31 | Boston Scientific Scimed, Inc. | Controlled delivery of therapeutic agents by insertable medical devices |
| EP1250164B1 (en) | 2000-01-24 | 2005-11-23 | Biocompatibles UK Limited | Coated implants |
| US7828835B2 (en) | 2000-03-01 | 2010-11-09 | Medinol Ltd. | Longitudinally flexible stent |
| US6379382B1 (en) | 2000-03-13 | 2002-04-30 | Jun Yang | Stent having cover with drug delivery capability |
| US20020007213A1 (en) | 2000-05-19 | 2002-01-17 | Robert Falotico | Drug/drug delivery systems for the prevention and treatment of vascular disease |
| US6776796B2 (en) | 2000-05-12 | 2004-08-17 | Cordis Corportation | Antiinflammatory drug and delivery device |
| US20020005206A1 (en) | 2000-05-19 | 2002-01-17 | Robert Falotico | Antiproliferative drug and delivery device |
| US8252044B1 (en) | 2000-11-17 | 2012-08-28 | Advanced Bio Prosthestic Surfaces, Ltd. | Device for in vivo delivery of bioactive agents and method of manufacture thereof |
| US6673385B1 (en) | 2000-05-31 | 2004-01-06 | Advanced Cardiovascular Systems, Inc. | Methods for polymeric coatings stents |
| AU6539101A (en) | 2000-06-05 | 2001-12-17 | G David Jang | Intravascular stent with increasing coating retaining capacity |
| US6723373B1 (en) | 2000-06-16 | 2004-04-20 | Cordis Corporation | Device and process for coating stents |
| US20020077693A1 (en) | 2000-12-19 | 2002-06-20 | Barclay Bruce J. | Covered, coiled drug delivery stent and method |
| US6540775B1 (en) | 2000-06-30 | 2003-04-01 | Cordis Corporation | Ultraflexible open cell stent |
| US6709451B1 (en) | 2000-07-14 | 2004-03-23 | Norman Noble, Inc. | Channeled vascular stent apparatus and method |
| US6555157B1 (en) | 2000-07-25 | 2003-04-29 | Advanced Cardiovascular Systems, Inc. | Method for coating an implantable device and system for performing the method |
| JP4841066B2 (en) | 2000-09-01 | 2011-12-21 | ライスユニバーシティ | Nitric oxide-forming hydrogel materials |
| AU2001288809A1 (en) | 2000-09-26 | 2002-04-08 | Advanced Cardiovascular Systems Inc. | A method of loading a substance onto an implantable device |
| US6716444B1 (en) | 2000-09-28 | 2004-04-06 | Advanced Cardiovascular Systems, Inc. | Barriers for polymer-coated implantable medical devices and methods for making the same |
| AU1129902A (en) | 2000-09-29 | 2002-04-08 | Cordis Corp | Coated medical devices |
| WO2002059261A2 (en) | 2000-12-07 | 2002-08-01 | The Medstar Research Institute | Inhibition of restenosis using a dna-coated stent |
| US20020082678A1 (en) * | 2000-12-22 | 2002-06-27 | Motasim Sirhan | Intravascular delivery of mizoribine |
| US7077859B2 (en) | 2000-12-22 | 2006-07-18 | Avantec Vascular Corporation | Apparatus and methods for variably controlled substance delivery from implanted prostheses |
| EP1223305B1 (en) | 2001-01-16 | 2008-04-23 | Services Petroliers Schlumberger | Bi-stable expandable device and method for expanding such a device |
| US6706274B2 (en) | 2001-01-18 | 2004-03-16 | Scimed Life Systems, Inc. | Differential delivery of nitric oxide |
| US20020127263A1 (en) | 2001-02-27 | 2002-09-12 | Wenda Carlyle | Peroxisome proliferator-acitvated receptor gamma ligand eluting medical device |
| US6780424B2 (en) | 2001-03-30 | 2004-08-24 | Charles David Claude | Controlled morphologies in polymer drug for release of drugs from polymer films |
| WO2002087586A1 (en) | 2001-04-26 | 2002-11-07 | Control Delivery Systems, Inc. | Sustained release drug delivery system containing codrugs |
| US6551352B2 (en) | 2001-05-03 | 2003-04-22 | Bionx Implants, Inc. | Method for attaching axial filaments to a self expanding stent |
| US7862495B2 (en) * | 2001-05-31 | 2011-01-04 | Advanced Cardiovascular Systems, Inc. | Radiation or drug delivery source with activity gradient to minimize edge effects |
| US20030050687A1 (en) | 2001-07-03 | 2003-03-13 | Schwade Nathan D. | Biocompatible stents and method of deployment |
| WO2003007842A2 (en) | 2001-07-18 | 2003-01-30 | Disa Vascular (Pty) Ltd | Stents |
| US20030077312A1 (en) | 2001-10-22 | 2003-04-24 | Ascher Schmulewicz | Coated intraluminal stents and reduction of restenosis using same |
| US20030088307A1 (en) | 2001-11-05 | 2003-05-08 | Shulze John E. | Potent coatings for stents |
| EP1310242A1 (en) | 2001-11-13 | 2003-05-14 | SORIN BIOMEDICA CARDIO S.p.A. | Carrier and kit for endoluminal delivery of active principles |
| US7014654B2 (en) | 2001-11-30 | 2006-03-21 | Scimed Life Systems, Inc. | Stent designed for the delivery of therapeutic substance or other agents |
| US20030181973A1 (en) | 2002-03-20 | 2003-09-25 | Harvinder Sahota | Reduced restenosis drug containing stents |
| EP1348402A1 (en) | 2002-03-29 | 2003-10-01 | Advanced Laser Applications Holding S.A. | Intraluminal endoprosthesis, radially expandable, perforated for drug delivery |
| US20030204239A1 (en) | 2002-04-26 | 2003-10-30 | Wenda Carlyle | Endovascular stent with a preservative coating |
| US6645547B1 (en) | 2002-05-02 | 2003-11-11 | Labcoat Ltd. | Stent coating device |
| US7332160B2 (en) | 2002-07-12 | 2008-02-19 | Boston Scientific Scimed, Inc. | Medical device and method for tissue removal and repair |
| US6702850B1 (en) | 2002-09-30 | 2004-03-09 | Mediplex Corporation Korea | Multi-coated drug-eluting stent for antithrombosis and antirestenosis |
| KR20130032407A (en) | 2002-11-08 | 2013-04-01 | 코너 메드시스템즈, 엘엘씨 | Method and apparatus for reducing tissue damage after ischemic injury |
-
2004
- 2004-05-27 US US10/857,201 patent/US20040254635A1/en not_active Abandoned
-
2007
- 2007-10-26 US US11/925,344 patent/US8361537B2/en not_active Expired - Fee Related
-
2009
- 2009-03-30 US US12/413,727 patent/US20090228095A1/en not_active Abandoned
Patent Citations (55)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20030036794A1 (en) * | 1995-06-07 | 2003-02-20 | Cook Incorporated | Coated implantable medical device |
| US5824049A (en) * | 1995-06-07 | 1998-10-20 | Med Institute, Inc. | Coated implantable medical device |
| US5873904A (en) * | 1995-06-07 | 1999-02-23 | Cook Incorporated | Silver implantable medical device |
| US5609629A (en) * | 1995-06-07 | 1997-03-11 | Med Institute, Inc. | Coated implantable medical device |
| US20030028244A1 (en) * | 1995-06-07 | 2003-02-06 | Cook Incorporated | Coated implantable medical device |
| US6096070A (en) * | 1995-06-07 | 2000-08-01 | Med Institute Inc. | Coated implantable medical device |
| US5992769A (en) * | 1995-06-09 | 1999-11-30 | The Regents Of The University Of Michigan | Microchannel system for fluid delivery |
| US5797898A (en) * | 1996-07-02 | 1998-08-25 | Massachusetts Institute Of Technology | Microchip drug delivery devices |
| US6123861A (en) * | 1996-07-02 | 2000-09-26 | Massachusetts Institute Of Technology | Fabrication of microchip drug delivery devices |
| US6071305A (en) * | 1996-11-25 | 2000-06-06 | Alza Corporation | Directional drug delivery stent and method of use |
| US6585764B2 (en) * | 1997-04-18 | 2003-07-01 | Cordis Corporation | Stent with therapeutically active dosage of rapamycin coated thereon |
| US20030176915A1 (en) * | 1997-04-18 | 2003-09-18 | Carol Wright | Local delivery of rapamycin for treatment of proliferative sequelae associated with PTCA procedures, including delivery using a modified stent |
| US20010027340A1 (en) * | 1997-04-18 | 2001-10-04 | Carol Wright | Stent with therapeutically active dosage of rapamycin coated thereon |
| US6273913B1 (en) * | 1997-04-18 | 2001-08-14 | Cordis Corporation | Modified stent useful for delivery of drugs along stent strut |
| US5972027A (en) * | 1997-09-30 | 1999-10-26 | Scimed Life Systems, Inc | Porous stent drug delivery system |
| US6273908B1 (en) * | 1997-10-24 | 2001-08-14 | Robert Ndondo-Lay | Stents |
| US6156062A (en) * | 1997-12-03 | 2000-12-05 | Ave Connaught | Helically wrapped interlocking stent |
| US20030199970A1 (en) * | 1998-03-30 | 2003-10-23 | Conor Medsystems, Inc. | Expandable medical device for delivery of beneficial agent |
| US6241762B1 (en) * | 1998-03-30 | 2001-06-05 | Conor Medsystems, Inc. | Expandable medical device with ductile hinges |
| US6562065B1 (en) * | 1998-03-30 | 2003-05-13 | Conor Medsystems, Inc. | Expandable medical device with beneficial agent delivery mechanism |
| US20040122505A1 (en) * | 1998-03-30 | 2004-06-24 | Conor Medsystems, Inc. | Expandable medical device with curved hinge |
| US20010029351A1 (en) * | 1998-04-16 | 2001-10-11 | Robert Falotico | Drug combinations and delivery devices for the prevention and treatment of vascular disease |
| US6299604B1 (en) * | 1998-08-20 | 2001-10-09 | Cook Incorporated | Coated implantable medical device |
| US20020032414A1 (en) * | 1998-08-20 | 2002-03-14 | Ragheb Anthony O. | Coated implantable medical device |
| US6730064B2 (en) * | 1998-08-20 | 2004-05-04 | Cook Incorporated | Coated implantable medical device |
| US20020028243A1 (en) * | 1998-09-25 | 2002-03-07 | Masters David B. | Protein matrix materials, devices and methods of making and using thereof |
| US6206915B1 (en) * | 1998-09-29 | 2001-03-27 | Medtronic Ave, Inc. | Drug storing and metering stent |
| US6293967B1 (en) * | 1998-10-29 | 2001-09-25 | Conor Medsystems, Inc. | Expandable medical device with ductile hinges |
| US6730116B1 (en) * | 1999-04-16 | 2004-05-04 | Medtronic, Inc. | Medical device for intraluminal endovascular stenting |
| US6379381B1 (en) * | 1999-09-03 | 2002-04-30 | Advanced Cardiovascular Systems, Inc. | Porous prosthesis and a method of depositing substances into the pores |
| US6656162B2 (en) * | 1999-11-17 | 2003-12-02 | Microchips, Inc. | Implantable drug delivery stents |
| US6537256B2 (en) * | 1999-11-17 | 2003-03-25 | Microchips, Inc. | Microfabricated devices for the delivery of molecules into a carrier fluid |
| US6491666B1 (en) * | 1999-11-17 | 2002-12-10 | Microchips, Inc. | Microfabricated devices for the delivery of molecules into a carrier fluid |
| US20030100865A1 (en) * | 1999-11-17 | 2003-05-29 | Santini John T. | Implantable drug delivery stents |
| US6423092B2 (en) * | 1999-12-22 | 2002-07-23 | Ethicon, Inc. | Biodegradable stent |
| US6551838B2 (en) * | 2000-03-02 | 2003-04-22 | Microchips, Inc. | Microfabricated devices for the storage and selective exposure of chemicals and devices |
| US20020007209A1 (en) * | 2000-03-06 | 2002-01-17 | Scheerder Ivan De | Intraluminar perforated radially expandable drug delivery prosthesis and a method for the production thereof |
| US20030216699A1 (en) * | 2000-05-12 | 2003-11-20 | Robert Falotico | Coated medical devices for the prevention and treatment of vascular disease |
| US6783543B2 (en) * | 2000-06-05 | 2004-08-31 | Scimed Life Systems, Inc. | Intravascular stent with increasing coating retaining capacity |
| US20020038145A1 (en) * | 2000-06-05 | 2002-03-28 | Jang G. David | Intravascular stent with increasing coating retaining capacity |
| US6254632B1 (en) * | 2000-09-28 | 2001-07-03 | Advanced Cardiovascular Systems, Inc. | Implantable medical device having protruding surface structures for drug delivery and cover attachment |
| US20020082680A1 (en) * | 2000-10-16 | 2002-06-27 | Shanley John F. | Expandable medical device for delivery of beneficial agent |
| US20020068969A1 (en) * | 2000-10-16 | 2002-06-06 | Shanley John F. | Expandable medical device with improved spatial distribution |
| US6506437B1 (en) * | 2000-10-17 | 2003-01-14 | Advanced Cardiovascular Systems, Inc. | Methods of coating an implantable device having depots formed in a surface thereof |
| US6558733B1 (en) * | 2000-10-26 | 2003-05-06 | Advanced Cardiovascular Systems, Inc. | Method for etching a micropatterned microdepot prosthesis |
| US6758859B1 (en) * | 2000-10-30 | 2004-07-06 | Kenny L. Dang | Increased drug-loading and reduced stress drug delivery device |
| US6635082B1 (en) * | 2000-12-29 | 2003-10-21 | Advanced Cardiovascular Systems Inc. | Radiopaque stent |
| US6752829B2 (en) * | 2001-01-30 | 2004-06-22 | Scimed Life Systems, Inc. | Stent with channel(s) for containing and delivering a biologically active material and method for manufacturing the same |
| US20020155212A1 (en) * | 2001-04-24 | 2002-10-24 | Hossainy Syed Faiyaz Ahmed | Coating for a stent and a method of forming the same |
| US6699281B2 (en) * | 2001-07-20 | 2004-03-02 | Sorin Biomedica Cardio S.P.A. | Angioplasty stents |
| US20030068355A1 (en) * | 2001-08-20 | 2003-04-10 | Shanley John F. | Therapeutic agent delivery device with protective separating layer |
| US20030060877A1 (en) * | 2001-09-25 | 2003-03-27 | Robert Falotico | Coated medical devices for the treatment of vascular disease |
| US20040127976A1 (en) * | 2002-09-20 | 2004-07-01 | Conor Medsystems, Inc. | Method and apparatus for loading a beneficial agent into an expandable medical device |
| US20040127977A1 (en) * | 2002-09-20 | 2004-07-01 | Conor Medsystems, Inc. | Expandable medical device with openings for delivery of multiple beneficial agents |
| US20040144506A1 (en) * | 2002-10-17 | 2004-07-29 | Bos Gmbh & Co. Kg | Window shade with extraction slot cover |
Cited By (103)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8313521B2 (en) | 1995-06-07 | 2012-11-20 | Cook Medical Technologies Llc | Method of delivering an implantable medical device with a bioabsorbable coating |
| US7892221B2 (en) | 1996-07-02 | 2011-02-22 | Massachusetts Institute Of Technology | Method of controlled drug delivery from implant device |
| US7070590B1 (en) | 1996-07-02 | 2006-07-04 | Massachusetts Institute Of Technology | Microchip drug delivery devices |
| US7918842B2 (en) | 1996-07-02 | 2011-04-05 | Massachusetts Institute Of Technology | Medical device with controlled reservoir opening |
| US8066763B2 (en) | 1998-04-11 | 2011-11-29 | Boston Scientific Scimed, Inc. | Drug-releasing stent with ceramic-containing layer |
| US8303643B2 (en) | 2001-06-27 | 2012-11-06 | Remon Medical Technologies Ltd. | Method and device for electrochemical formation of therapeutic species in vivo |
| US7842083B2 (en) | 2001-08-20 | 2010-11-30 | Innovational Holdings, Llc. | Expandable medical device with improved spatial distribution |
| US7951392B2 (en) | 2002-08-16 | 2011-05-31 | Boston Scientific Scimed, Inc. | Microarray drug delivery coatings |
| US20110017346A1 (en) * | 2002-09-20 | 2011-01-27 | Innovational Holdings, Llc | Method and apparatus for loading a beneficial agent into an expandable medical device |
| US9498358B2 (en) | 2002-09-20 | 2016-11-22 | Innovational Holdings Llc | Implantable medical device with openings for delivery of beneficial agents with combination release kinetics |
| US9254202B2 (en) | 2002-09-20 | 2016-02-09 | Innovational Holdings Llc | Method and apparatus for loading a beneficial agent into an expandable medical device |
| US8349390B2 (en) | 2002-09-20 | 2013-01-08 | Conor Medsystems, Inc. | Method and apparatus for loading a beneficial agent into an expandable medical device |
| US20050100582A1 (en) * | 2003-11-06 | 2005-05-12 | Stenzel Eric B. | Method and apparatus for controlled delivery of active substance |
| US7435256B2 (en) * | 2003-11-06 | 2008-10-14 | Boston Scientific Scimed, Inc. | Method and apparatus for controlled delivery of active substance |
| US8501213B2 (en) | 2004-03-19 | 2013-08-06 | Abbott Laboratories | Multiple drug delivery from a balloon and a prosthesis |
| US8956639B2 (en) | 2004-03-19 | 2015-02-17 | Abbott Laboratories | Multiple drug delivery from a balloon and prosthesis |
| JP2008532692A (en) * | 2005-03-14 | 2008-08-21 | コナー・ミッドシステムズ・インコーポレイテッド | Expanded medical device with an opening for delivering multiple active substances |
| EP1858442A4 (en) * | 2005-03-14 | 2010-09-01 | Conor Medsystems Inc | Expandable medical device with openings for delivery of multiple beneficial agents |
| EP1858442A2 (en) * | 2005-03-14 | 2007-11-28 | Conor Medsystems, Inc. | Expandable medical device with openings for delivery of multiple beneficial agents |
| EP1890648A2 (en) * | 2005-06-06 | 2008-02-27 | Innovational Holdings, LLC | Implantable medical device with openings for delivery of beneficial agents with combination release kinetics |
| EP1890648A4 (en) * | 2005-06-06 | 2012-05-02 | Innovational Holdings Llc | Implantable medical device with openings for delivery of beneficial agents with combination release kinetics |
| US20070055352A1 (en) * | 2005-09-07 | 2007-03-08 | Wendy Naimark | Stent with pockets for containing a therapeutic agent |
| WO2007030534A2 (en) | 2005-09-07 | 2007-03-15 | Boston Scientific Limited | Stent with pockets containing a therapeutic agent |
| WO2007030534A3 (en) * | 2005-09-07 | 2007-05-03 | Boston Scient Scimed Inc | Stent with pockets containing a therapeutic agent |
| US8840660B2 (en) | 2006-01-05 | 2014-09-23 | Boston Scientific Scimed, Inc. | Bioerodible endoprostheses and methods of making the same |
| US8089029B2 (en) | 2006-02-01 | 2012-01-03 | Boston Scientific Scimed, Inc. | Bioabsorbable metal medical device and method of manufacture |
| US20100233350A1 (en) * | 2006-03-15 | 2010-09-16 | Boston Scientific Scimed, Inc. | Drug delivery composition and methods of making same using nanofabrication |
| WO2007108916A3 (en) * | 2006-03-15 | 2007-12-27 | Boston Scient Scimed Inc | Drug delivery composition and methods of making same using nanofabrication |
| WO2007108916A2 (en) * | 2006-03-15 | 2007-09-27 | Boston Scientific Scimed, Inc. | Drug delivery composition and methods of making same using nanofabrication |
| US8574615B2 (en) | 2006-03-24 | 2013-11-05 | Boston Scientific Scimed, Inc. | Medical devices having nanoporous coatings for controlled therapeutic agent delivery |
| US8187620B2 (en) | 2006-03-27 | 2012-05-29 | Boston Scientific Scimed, Inc. | Medical devices comprising a porous metal oxide or metal material and a polymer coating for delivering therapeutic agents |
| US8048150B2 (en) | 2006-04-12 | 2011-11-01 | Boston Scientific Scimed, Inc. | Endoprosthesis having a fiber meshwork disposed thereon |
| US8815275B2 (en) | 2006-06-28 | 2014-08-26 | Boston Scientific Scimed, Inc. | Coatings for medical devices comprising a therapeutic agent and a metallic material |
| US8771343B2 (en) | 2006-06-29 | 2014-07-08 | Boston Scientific Scimed, Inc. | Medical devices with selective titanium oxide coatings |
| US8052743B2 (en) | 2006-08-02 | 2011-11-08 | Boston Scientific Scimed, Inc. | Endoprosthesis with three-dimensional disintegration control |
| US8353949B2 (en) | 2006-09-14 | 2013-01-15 | Boston Scientific Scimed, Inc. | Medical devices with drug-eluting coating |
| US8057534B2 (en) | 2006-09-15 | 2011-11-15 | Boston Scientific Scimed, Inc. | Bioerodible endoprostheses and methods of making the same |
| US8052744B2 (en) | 2006-09-15 | 2011-11-08 | Boston Scientific Scimed, Inc. | Medical devices and methods of making the same |
| US8128689B2 (en) | 2006-09-15 | 2012-03-06 | Boston Scientific Scimed, Inc. | Bioerodible endoprosthesis with biostable inorganic layers |
| US8808726B2 (en) | 2006-09-15 | 2014-08-19 | Boston Scientific Scimed. Inc. | Bioerodible endoprostheses and methods of making the same |
| US8002821B2 (en) | 2006-09-18 | 2011-08-23 | Boston Scientific Scimed, Inc. | Bioerodible metallic ENDOPROSTHESES |
| US7981150B2 (en) | 2006-11-09 | 2011-07-19 | Boston Scientific Scimed, Inc. | Endoprosthesis with coatings |
| US8715339B2 (en) | 2006-12-28 | 2014-05-06 | Boston Scientific Scimed, Inc. | Bioerodible endoprostheses and methods of making the same |
| US8080055B2 (en) | 2006-12-28 | 2011-12-20 | Boston Scientific Scimed, Inc. | Bioerodible endoprostheses and methods of making the same |
| US8070797B2 (en) | 2007-03-01 | 2011-12-06 | Boston Scientific Scimed, Inc. | Medical device with a porous surface for delivery of a therapeutic agent |
| US8431149B2 (en) | 2007-03-01 | 2013-04-30 | Boston Scientific Scimed, Inc. | Coated medical devices for abluminal drug delivery |
| AU2008201395B2 (en) * | 2007-03-28 | 2013-08-15 | Cardinal Health 529, Llc | Short term sustained drug-delivery system for implantable medical devices and method of making the same |
| US20080243241A1 (en) * | 2007-03-28 | 2008-10-02 | Zhao Jonathon Z | Short term sustained drug-delivery system for implantable medical devices and method of making the same |
| US8067054B2 (en) | 2007-04-05 | 2011-11-29 | Boston Scientific Scimed, Inc. | Stents with ceramic drug reservoir layer and methods of making and using the same |
| US7976915B2 (en) | 2007-05-23 | 2011-07-12 | Boston Scientific Scimed, Inc. | Endoprosthesis with select ceramic morphology |
| US7942926B2 (en) | 2007-07-11 | 2011-05-17 | Boston Scientific Scimed, Inc. | Endoprosthesis coating |
| US8002823B2 (en) | 2007-07-11 | 2011-08-23 | Boston Scientific Scimed, Inc. | Endoprosthesis coating |
| US9284409B2 (en) | 2007-07-19 | 2016-03-15 | Boston Scientific Scimed, Inc. | Endoprosthesis having a non-fouling surface |
| WO2009014692A1 (en) * | 2007-07-24 | 2009-01-29 | Boston Scientific Limited | Stents with polymer-free coatings for delivering a therapeutic agent |
| US7931683B2 (en) | 2007-07-27 | 2011-04-26 | Boston Scientific Scimed, Inc. | Articles having ceramic coated surfaces |
| US8815273B2 (en) | 2007-07-27 | 2014-08-26 | Boston Scientific Scimed, Inc. | Drug eluting medical devices having porous layers |
| US8221822B2 (en) | 2007-07-31 | 2012-07-17 | Boston Scientific Scimed, Inc. | Medical device coating by laser cladding |
| US8900292B2 (en) | 2007-08-03 | 2014-12-02 | Boston Scientific Scimed, Inc. | Coating for medical device having increased surface area |
| US8052745B2 (en) | 2007-09-13 | 2011-11-08 | Boston Scientific Scimed, Inc. | Endoprosthesis |
| WO2009055720A1 (en) | 2007-10-26 | 2009-04-30 | University Of Virginia Patent Foundation | System for treatment and imaging using ultrasonic energy and microbubbles and related method thereof |
| US20100331686A1 (en) * | 2007-10-26 | 2010-12-30 | Hossack John A | System for Treatment and Imaging Using Ultrasonic Energy and Microbubbles and Related Method Thereof |
| US9526922B2 (en) | 2007-10-26 | 2016-12-27 | University Of Virginia Patent Foundation | System for treatment and imaging using ultrasonic energy and microbubbles and related method thereof |
| US9895158B2 (en) | 2007-10-26 | 2018-02-20 | University Of Virginia Patent Foundation | Method and apparatus for accelerated disintegration of blood clot |
| US8622911B2 (en) | 2007-10-26 | 2014-01-07 | University Of Virginia Patent Foundation | System for treatment and imaging using ultrasonic energy and microbubbles and related method thereof |
| US10893881B2 (en) | 2007-10-26 | 2021-01-19 | University Of Virginia Patent Foundation | Method and apparatus for accelerated disintegration of blood clot |
| US8216632B2 (en) | 2007-11-02 | 2012-07-10 | Boston Scientific Scimed, Inc. | Endoprosthesis coating |
| US7938855B2 (en) | 2007-11-02 | 2011-05-10 | Boston Scientific Scimed, Inc. | Deformable underlayer for stent |
| US8029554B2 (en) | 2007-11-02 | 2011-10-04 | Boston Scientific Scimed, Inc. | Stent with embedded material |
| WO2009079132A1 (en) * | 2007-12-19 | 2009-06-25 | Boston Scientific Scimed, Inc. | Stent |
| US7722661B2 (en) | 2007-12-19 | 2010-05-25 | Boston Scientific Scimed, Inc. | Stent |
| US20090163991A1 (en) * | 2007-12-19 | 2009-06-25 | Boston Scientific Scimed, Inc. | Stent |
| US20100222868A1 (en) * | 2007-12-19 | 2010-09-02 | Boston Scientific Scimed, Inc. | Stent |
| US7922756B2 (en) | 2007-12-19 | 2011-04-12 | Boston Scientific Scimed, Inc. | Stent |
| US20110190873A1 (en) * | 2007-12-19 | 2011-08-04 | Boston Scientific Scimed, Inc. | Stent |
| US8920491B2 (en) | 2008-04-22 | 2014-12-30 | Boston Scientific Scimed, Inc. | Medical devices having a coating of inorganic material |
| US8932346B2 (en) | 2008-04-24 | 2015-01-13 | Boston Scientific Scimed, Inc. | Medical devices having inorganic particle layers |
| WO2009135008A3 (en) * | 2008-05-01 | 2010-12-02 | Boston Scientific Scimed, Inc. | Drug-loaded medical devices and methods for manufacturing drug-loaded medical devices |
| WO2009135008A2 (en) * | 2008-05-01 | 2009-11-05 | Boston Scientific Scimed, Inc. | Drug-loaded medical devices and methods for manufacturing drug-loaded medical devices |
| US20090274740A1 (en) * | 2008-05-01 | 2009-11-05 | Boston Scientific Scimed, Inc. | Drug-loaded medical devices and methods for manufacturing drug-loaded medical devices |
| US7998192B2 (en) | 2008-05-09 | 2011-08-16 | Boston Scientific Scimed, Inc. | Endoprostheses |
| US8236046B2 (en) | 2008-06-10 | 2012-08-07 | Boston Scientific Scimed, Inc. | Bioerodible endoprosthesis |
| US20090319032A1 (en) * | 2008-06-18 | 2009-12-24 | Boston Scientific Scimed, Inc | Endoprosthesis coating |
| US8449603B2 (en) * | 2008-06-18 | 2013-05-28 | Boston Scientific Scimed, Inc. | Endoprosthesis coating |
| US20100010621A1 (en) * | 2008-07-11 | 2010-01-14 | Biotronik Vi Patent Ag | Stent having biodegradable stent struts and drug depots |
| US7985252B2 (en) | 2008-07-30 | 2011-07-26 | Boston Scientific Scimed, Inc. | Bioerodible endoprosthesis |
| US20100028403A1 (en) * | 2008-07-31 | 2010-02-04 | Boston Scientific Scimed, Inc. | Medical devices for therapeutic agent delivery |
| US8642063B2 (en) | 2008-08-22 | 2014-02-04 | Cook Medical Technologies Llc | Implantable medical device coatings with biodegradable elastomer and releasable taxane agent |
| US8382824B2 (en) | 2008-10-03 | 2013-02-26 | Boston Scientific Scimed, Inc. | Medical implant having NANO-crystal grains with barrier layers of metal nitrides or fluorides |
| US8231980B2 (en) | 2008-12-03 | 2012-07-31 | Boston Scientific Scimed, Inc. | Medical implants including iridium oxide |
| US8267992B2 (en) | 2009-03-02 | 2012-09-18 | Boston Scientific Scimed, Inc. | Self-buffering medical implants |
| US8071156B2 (en) | 2009-03-04 | 2011-12-06 | Boston Scientific Scimed, Inc. | Endoprostheses |
| US8287937B2 (en) | 2009-04-24 | 2012-10-16 | Boston Scientific Scimed, Inc. | Endoprosthese |
| US8951595B2 (en) * | 2009-12-11 | 2015-02-10 | Abbott Cardiovascular Systems Inc. | Coatings with tunable molecular architecture for drug-coated balloon |
| US20110143014A1 (en) * | 2009-12-11 | 2011-06-16 | John Stankus | Coatings with tunable molecular architecture for drug-coated balloon |
| US8480620B2 (en) | 2009-12-11 | 2013-07-09 | Abbott Cardiovascular Systems Inc. | Coatings with tunable solubility profile for drug-coated balloon |
| US8668732B2 (en) | 2010-03-23 | 2014-03-11 | Boston Scientific Scimed, Inc. | Surface treated bioerodible metal endoprostheses |
| US11752698B2 (en) * | 2012-01-24 | 2023-09-12 | Smith & Nephew, Inc. | Porous structure and methods of making same |
| US20210308948A1 (en) * | 2012-01-24 | 2021-10-07 | Smith & Nephew, Inc. | Porous structure and methods of making same |
| EP2727561A1 (en) * | 2012-10-31 | 2014-05-07 | Cook Medical Technologies LLC | Fixation process for nesting stents |
| US20140121750A1 (en) * | 2012-10-31 | 2014-05-01 | Cook Medical Technologies Llc | Fixation Process For Nesting Stents |
| US10786372B2 (en) * | 2013-04-24 | 2020-09-29 | Vascular Dynamics, Inc. | Implantable vascular device having longitudinal struts |
| CN107456301A (en) * | 2013-04-24 | 2017-12-12 | 血管动力公司 | Implantable vascular devices with longitudinal holding pole |
| US20160038317A1 (en) * | 2013-04-24 | 2016-02-11 | Vascular Dynamics, Inc. | Implantable vascular device having longitudinal struts |
Also Published As
| Publication number | Publication date |
|---|---|
| US20090228095A1 (en) | 2009-09-10 |
| US8361537B2 (en) | 2013-01-29 |
| US20080097581A1 (en) | 2008-04-24 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US8361537B2 (en) | Expandable medical device with beneficial agent concentration gradient | |
| EP1420719B1 (en) | Expandable medical device for delivery of beneficial agent | |
| EP1768610B1 (en) | Expandable medical device for treating cardiac arrhythmias | |
| EP1749544B1 (en) | Therapeutic agent delivery device with protective separating layer | |
| AU2010200882B2 (en) | Expandable medical device for delivery of beneficial agent | |
| AU2002310295A1 (en) | Expandable medical device for delivery of beneficial agent |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AS | Assignment |
Owner name: CONOR MEDSYSTEMS, INC., CALIFORNIA Free format text: ASSIGNMENT OF ASSIGNORS INTEREST;ASSIGNORS:SHANLEY, JOHN F.;EIGLER, NEAL L.;REEL/FRAME:015691/0763 Effective date: 20040811 |
|
| AS | Assignment |
Owner name: INNOVATIONAL HOLDINGS LLC, NEW JERSEY Free format text: ASSIGNMENT OF ASSIGNORS INTEREST;ASSIGNOR:CONOR MEDSYSTEMS, INC.;REEL/FRAME:019955/0487 Effective date: 20070306 Owner name: INNOVATIONAL HOLDINGS LLC,NEW JERSEY Free format text: ASSIGNMENT OF ASSIGNORS INTEREST;ASSIGNOR:CONOR MEDSYSTEMS, INC.;REEL/FRAME:019955/0487 Effective date: 20070306 |
|
| AS | Assignment |
Owner name: INNOVATIONAL HOLDINGS LLC, NEW JERSEY Free format text: ASSIGNMENT OF ASSIGNORS INTEREST;ASSIGNOR:CONOR MEDSYSTEMS, INC.;REEL/FRAME:023538/0021 Effective date: 20070306 Owner name: INNOVATIONAL HOLDINGS LLC,NEW JERSEY Free format text: ASSIGNMENT OF ASSIGNORS INTEREST;ASSIGNOR:CONOR MEDSYSTEMS, INC.;REEL/FRAME:023538/0021 Effective date: 20070306 |
|
| STCB | Information on status: application discontinuation |
Free format text: ABANDONED -- FAILURE TO RESPOND TO AN OFFICE ACTION |